Did my EcoRI restriction digestion work? picture included! - (Jun/27/2012 )
Sorry one more thing, I am also working on another gene at the same time. I follow the exact same steps as the other gene I asked my questions about. For the second gene I recently ligated it into the TOPO vector as well. I analyzed a few colonies after transformation for positive clones. I attached a picture of my colony PCR showing positive transformants, my insert size is 585bp. Based on that, it seems the picture indicates that all my transformants are positive.

Like the other gene I needed to double digest the TOPO vector+insert using NdeI and XhoI enzymes to later ligate into pET14b vector that is double digested with NdeI and XhoI enzymes as well.
I attached a photo of my TOPO vector+ insert double digest which shows a weird banding pattern, what could the reason be for this unwanted banding pattern?
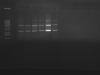
Thanks for all your help 
I think that you have to digest more template to make sure you cann see your 600 bp fragment in the gel. Also the staining is not good. You can barely see the ladder, especially the small sizes.
I don't know the expected sizes and the shown one, but what could also have happened is that your digestion was not complete. It seems a little bit that only one enzyme has cut (which I cannot exactly say because of missing sizes).Also showing the undigested vector might help.
Just give it another try. You can check with another enzymes (e.g. EcoRI) at the same time to make sure your construct is ok. It once happened to me that the fragement was double ligated into the vector...
Hi Papaver,
Thanks for your reply. I will take into consideration your ideas. But do you think there is a problem with my double digestion protocol listed below
TOPO+insert 1 (concentration 44.2ng/uL)
Plasmid DNA 3ug (67.9uL)
10X buffer H 3uL
NdeI 1uL
XhoI 3uL
NO WATER ADDED
TOPO+insert 2 (concentration 43.3ng/uL)
Plasmid DNA 3ug (69.3uL)
10X buffer H 3uL
NdeI 1uL
XhoI 1uL
NO WATER ADDED
Why are you adding only 3ul of 10x buffer?  According to your recipe, the final concentration of your restriction buffer is 0.4x which is a 60% lower than it should be, so yes there is a problem with your reactions, as your buffering conditions will not be the optimal for the enzymes.
According to your recipe, the final concentration of your restriction buffer is 0.4x which is a 60% lower than it should be, so yes there is a problem with your reactions, as your buffering conditions will not be the optimal for the enzymes.
You need to dilute your 10x buffer to a final 1x buffer concentration, not just add 3µl per reaction (I don't understand where the 3ul come from). If you need to add that much volume of DNA, you can prepare the reactions in a final volume of 100ul, add 10ul of buffer, keep your DNA and enzyme volumes as they are, and then top up with water.
Just my 2 cents.
on the other hand, if there's an actual reason to have the buffer at 0.4x concentration just ignore my message 
Your 2 cents are worthy to me..hehe..
No, the 0.4x is not part of the reaction conditions. I think that is a dumb mistake by me. I am suppose to follow the general reaction conditions for restriction digestion which agrees with your suggestion.
I will definately give that protocol a try and hope for the best.
Thanks, you guys are awesome!
You should be adding water to your restriction digest. I'd recommend this reaction setup:
5 ul of a 10x buffer (use the right one, probably buffer 4 if you use NEB enzymes)
0.5 ul 100x BSA
1 ul each enzyme
1 ug of DNA (less than 15 ul total volume)
sufficient water to bring the volume to 50 ul (more than 27 ul)
This does several things:
1) correct final buffer concentration (1x)
2) dilutes impurities in your DNA prep that can cause problems (primarily ethanol and Gu-HCl)
3) reduces the concentration of glycerol in the reaction (REs are in 50% glycerol, and you need to keep the final concentration below 5%)
Your reaction will be mostly done in 30 minutes of incubation at 37, and if you choose your enzymes, you can heat kill it at 80 for 20 minutes and use it directly in a ligation. It's easiest to do this in a PCR cycler.
It is a mistake to try to maintain a high concentration of DNA in these reactions. You don't need it for ligation, and trying to do it leads to many problems. Load 20 ul of the cut product on a gel, use 2 ul in a ligation, and keep the rest in the freezer if you want.
Hey Phage 434,
Wow, thanks to you too, I will be honest with you a lot of the points you mentioned in your post I am not familiar with. It really helps to know these key points you mentioned for future lab work. I will also try your method and hope for the best result that are satisfying.
I really hope this forum keeps going, it is great help to beginners like me.
Hello All,
Almost a doctor, I would have tried your method as well but realized I did not have enough plasmid mini prep left  . I went ahead and tried only Phage434's latest suggested protocol. Unfortuantely, the digestion failed again. Here is a picture of how the digestion banding pattern is
. I went ahead and tried only Phage434's latest suggested protocol. Unfortuantely, the digestion failed again. Here is a picture of how the digestion banding pattern is 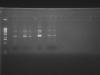
I forgot to run an undigested TOPO+insert on the same gel, and ended up running it by itself on a separate gel. Here is the image of the undigested TOPO+insert 
The image is not very clear, sorry.
Based on the image, I would assume that my samples did not get digested. I thought my enzymes are the problem, but one of my colleagues in the lab recently used the enzymes and their digestion was successfull.
I am confused as to what my problem is
Thanks again to all
So, I would say there is no insert.
Which clone(s) have you checked by digestion? Based on your PCR-Gel, I would have chosen samples 6 and 12.
One question considering your PCR...have you run a negative control? All bands besides lane 6 and 12 seem to me like they could be false positive.
Have you tried another enzyme just for checking your constructs? Are you sure that neither NdeI or XhoI cut in your insert?
I agree with Papaver. Try cutting with another enzyme, one that cuts your vector backbone. The bands look uncut. Are you expecting the NdeI and XhoI sites to be present in the original vector, or are they on your insert? It might be time for sequencing if you can't figure out what is happening. Dead enzyme would also explain this, so check that you can cut something else (or show that the DNA can be cut with a different enzyme).