Did my EcoRI restriction digestion work? picture included! - (Jun/27/2012 )
Hi Fluffy,
so first...if you buffer is 5 x , you would use 2 µl ; final concentrations should always be 1 x. You should also check if the buffer contains ATP (ligation reaction depends on it), otherwise you have to add it to the reaction. If the buffer is as old (and often reused) as the enzyme it might be better to add ATP.
Do you incubate your transformation mix prior to heat shock? If not, do this for around 30 min. After heat shock, add ~ 1 ml LB medium and incubate for at least one hour. I often incubate two. Try different amounts for plating...say 50 µl, 100 µl, 200 µl. Then pellet the remaining cells, remove a bit of LB and resupend the pellet in the remaining ~ 150 µl of LB. So you would plate all your cells. But I would do these "plating series" only for your ligation reaction 
Considering your colonies you got with the DD-plasmid...maybe some plasmids weren't double digested in your reaction and so they self ligated...if there are only 5 I would not worry...it can always happen that you get colonies with empty plasmid even if you do a double-digestion cloning.
Cheers
Thanks Papaver for your reply.
I believe the buffer has ATP as one of its ingredients and it is as old as the enzyme. I am thinking of getting a new one Friday ( an enzyme with a new buffer to be extra careful, I might get the 10X so that way I will stick to the 1.5uL)
Also for my transformation I do incubate it on ice for 5 min before heat shock. After heat shock I incubate again on ice for 2 min. Also according to my protocol after that I add 80uL SOC medium to ensure better spreading, and then I put on 37 at 250rpm speed for 1 hour. Thought I should be more clear 
So if I add SOC medium do you think I should still add LB medium ( I am guessing it has to have ampicillin antibiotic)?
Thanks again dear!
Also I forgot to mention for my reactions I incubated in a thermocycler at 16C overnight
Fluffy on Thu Jul 19 15:42:35 2012 said:
Thanks Papaver for your reply.
I believe the buffer has ATP as one of its ingredients and it is as old as the enzyme. I am thinking of getting a new one Friday ( an enzyme with a new buffer to be extra careful, I might get the 10X so that way I will stick to the 1.5uL)
Also for my transformation I do incubate it on ice for 5 min before heat shock. After heat shock I incubate again on ice for 2 min. Also according to my protocol after that I add 80uL SOC medium to ensure better spreading, and then I put on 37 at 250rpm speed for 1 hour. Thought I should be more clear
So if I add SOC medium do you think I should still add LB medium ( I am guessing it has to have ampicillin antibiotic)?
Thanks again dear!
SOC Medium is even better than LB, and for both: no antibiotics before plating the cells on LB/Amp-plates.
You can always play a bit around with your transformation mix. Incubation longer on ice before heat shock would give you cells more time to "get in contact" with the DNA. You can also add more of your ligation mix...1 µl is a small amount.
You can also plate more...
Hello Papaver
I proceeded with ligations and transformations. Your suggestions made things easier for me. Thanks . I did colony PCR for the chosen colonies from ligation plates to check for recombinant plasmids with my insert ligated. The result is the following attached picture: The PCR involved the use of primers specific to my insert and it seems to be my correct insert size: 447bp long.
The PCR involved the use of primers specific to my insert and it seems to be my correct insert size: 447bp long.
The next step my professor asked me to do a PCR to determine the correct insert orientation. I am not sure how this works and how PCR can be used to determine insert orientation. I understand that I have to use one primer specific to my vector (pET14b) located near the multiple cloning site, and a second primer specific to my insert. But I do not understand how it works.
I already designed my primers that are specific to my vector, I designed more than one.
Examples:
5'
5'
Do you have an idea about this method?
Thanks again,
Fluffy:)
For PCR to work, the two primers need to be facing one another. Depending on the orientation of your insert, a primer on the vector and another on the insert will either face each other or be oriented in the same direction. You can determine the orientation of the insert by checking which of these two PCR reactions work. You could also do a similar check by restriction digest, if you can find one (or possibly a collection) of RE sites on your vector and insert which give distinct patterns on digestion.
Hi Fluffy
since you have cloned your fragment into the vector using two different enzymes, the direction of the insert is given by the restriction sites. I suppose you have added the Nde site to the 5 prime of your insert and the Xho I site at the 3 prime end. Anything else would not make sense to me and that's why I would check a few plasmids isolated from your clones by restriction digest phage 434 has already mentioned. Besides this I would also sequence your construct to make sure your insert (or protein) is in frame.
Hi,
Hope everyone is well.
I did a PCR to confirm the orientation of my insert in the pET14b vector. I ran two PCRs, first using a vector primer and a forward primer of my insert, then the same vector primer and the reverse primer of my insert.
The PCR using my vector primer and forward primer of my insert should give me the correct orientation of my insert only if I get a band of size 600-700bp. I attached a gel image showing bands of approximatly that size.

The PCR using vector primer and the reverse primer of my insert gave me a weird pattern
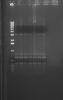
What do you think? I would assume my insert is in the correct orientation since the first picture gave clear bright bands.
Just a reminder the vector primer is a forward primer that was designed approx. 200bp-300bp of the multiple cloning site.
Should I move on?
Thanks again for all help!
I agree, your insert is in the direction amplified by your forward primer. The weird bands are trash products resulting from long extention times and low annealing temperatures priming to who knows what.
Sounds good Phage434
I will proceed with my work then!
Thanks again