olligo annealing for shRNA cloning - (May/26/2009 )
Bomber on Jun 4 2009, 10:01 PM said:
that looks weird.
One thing you might check, is the purity of your primers. In case you have a lot of degradation products left in your synthesized oligos it might hamper effective annealing.
Hi Bomber,
How can I check the purity of my primers? Thank you.
Functional Screens on Jun 5 2009, 09:33 AM said:
Hi Functinal Screens,
I didn't do any cloning but I indeed procceed to transformation, however, there's no colony observed though.
Well, when I ordered oligos for shRNA cloning the company where we ususally order primers suggested to have the oligos rather well purified; higher than any standard primer we usually order, since otherwise a lot of degradation products remain in the reaction.
This is also stated in the Ambion manual for shRNA cloning, so it seems to be critical point.
I don't know if this relevant but maybe this is why annelaing is hampered in your reaction. Though it is strange that you do not obtain any colonies.
Are you certain that your double digest works fine? I assume the restriction sites are in close proximity?
Have you succesfully cloned anything else successfully into the plasmid, like a negative control shRNA?
Hi,
I used the following protocol with good results:
Dilute oligo to 1µg/µl. 1 µl top oligo + 1 µl bottom oligo + 2 µl annealing buffer (10 x buffer: 100 mM Tris pH8,5; 1 M NaCl; 10 mM EDTA) + 16 µl ddH2O. Heat for 5 min in PCR cycler to 95°C. switch of and let cool to room temperature for 3 hours. use 2 µl of the annealing mix and ligate with 100 ng digested vector in 20 µl reaction volume over night. Electroporate 1 µl of the mixture in DH10B cells and plate.
To check annealing I usually run 1 µl of the annealing mix on a 3 % gel. I also often get 2 bands in the unannealed oligo control but it still works if the upper band in annealed oligo is stronger than in the untreated oligo.
Stardust
Bomber on Jun 5 2009, 04:27 PM said:
This is also stated in the Ambion manual for shRNA cloning, so it seems to be critical point.
I don't know if this relevant but maybe this is why annelaing is hampered in your reaction. Though it is strange that you do not obtain any colonies.
Are you certain that your double digest works fine? I assume the restriction sites are in close proximity?
Have you succesfully cloned anything else successfully into the plasmid, like a negative control shRNA?
I assume that my dual digestion for the cloning vector works well as we indeed saw the desired band as suggested by the protocol after running the gel. However, I've repeating the dual digestion again today. Hopefully it will yields some results. For the time being, we didn't do other cloning besides this one.
Thanks for your help a lot.

stardust on Jun 5 2009, 08:33 PM said:
I used the following protocol with good results:
Dilute oligo to 1µg/µl. 1 µl top oligo + 1 µl bottom oligo + 2 µl annealing buffer (10 x buffer: 100 mM Tris pH8,5; 1 M NaCl; 10 mM EDTA) + 16 µl ddH2O. Heat for 5 min in PCR cycler to 95°C. switch of and let cool to room temperature for 3 hours. use 2 µl of the annealing mix and ligate with 100 ng digested vector in 20 µl reaction volume over night. Electroporate 1 µl of the mixture in DH10B cells and plate.
To check annealing I usually run 1 µl of the annealing mix on a 3 % gel. I also often get 2 bands in the unannealed oligo control but it still works if the upper band in annealed oligo is stronger than in the untreated oligo.
Stardust
Thanks stardust. I'll try this protocol soon. Haha. But there is something that I would like to enquire first, when u turn off the PCR cycler, did u take out the oligo mixture to RT? Or you just keep it inside the PCR cycler? Then what about your ligation condition? 16C overnight incubation?
Thanks again.

Hi,
I just switch the machine off and take the oligos out after 3 h. I use 2 µl of the annealed oligo, 2 µl ligase buffer, 1µl T4 DNA ligase, 100 ng vector and fill up to 20 µl with ddH2O. I ligate 1 h at RT, over night at 4°C. Then I electroporate 1 µl in electrocomp. cells.
Stardust
stardust on Jun 8 2009, 08:15 PM said:
I just switch the machine off and take the oligos out after 3 h. I use 2 µl of the annealed oligo, 2 µl ligase buffer, 1µl T4 DNA ligase, 100 ng vector and fill up to 20 µl with ddH2O. I ligate 1 h at RT, over night at 4°C. Then I electroporate 1 µl in electrocomp. cells.
Stardust
Thanks lots!

I found this thread because I'm having difficulty subcloning into pLKO.1, and after trying the suggestions offered here, it still doesn't work.
I am not getting any colonies after ligation. I'm fairly certain that the vector is ok, as I've tried digesting it with various enzymes, and it gives the expected band pattern.
I decided to check whether the oligo annealing step was working, and ran the annealing reaction on a 3% gel, and...uh oh. I am attaching a picture of the agarose gel.
"O" is the single-stranded oligo. Lanes #1-5 are five separate annealing reactions. Group A reactions were done in a water bath (4 min at 95C, then let cool to room temp), and Group B were done in a PCR cycler (10 min at 70C, then decrease by 1C over the course of four hours until 25C). I used Addgene's annealing protocol (i.e. NEB's buffer 2, 20 uM of each oligo). I also tried stardust's protocol (10x annealing buffer, 1ug of each oligo), and after running it on a gel, see the same band pattern.
Each single-stranded oligo that I used is 58bp long. I'm not sure why there are three (!) bands for the two ss oligos on the gel, although the most prominent (middle) band appears to be the correct size. And then the annealed oligos...what's going on there?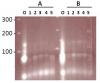
I also have similar problems that the single stranded and annealed mixtures look the same. I think this is probably because the primers can anneal to themselves as well as their complements. While they won't anneal across their whole length because the mid loop section and the ends are not complementary, the siRNA sequence itself, whether it's 21 or 28 bases long is able to dimerize none-the-less. The idea of slow cooling the annealing reaction is presumably so the mixture spends as long as possible between 70 and 80 degrees where the correct dimer alone should be able to form (no secondary structure or incorrect primer dimers will be stable here). Once the temperature drops below this however then the individual primers will be free to anneal intra and inter molecularly. The longer the complementary section (I'm trying 28-mer) the bigger this problem will be as there is relatively less difference between the forward and reverse primers.
Having rationalized like this however, I'm still not having any luck! I'm making attempt number 6 today....