amplified proviral genome and got human sequence - totally different sequences from what i expected (Mar/30/2010 )
Step 1: Create a PCR reaction with twice your normal volume. Split it in half. Cycle half. Run a gel with a marker lane, uncycled and cycled reactions in adjacent wells.
Your uncycled lane should show no bands whatsoever, because it should have sufficiently low template concentration that it is not visible.
HomeBrew on Apr 2 2010, 02:12 AM said:
gyma on Apr 1 2010, 08:06 AM said:
Yes, but your evidence suggests these bands are *not* amplification products -- if they were, they would (by definition) have the primer sequences in them. Thus, if they're not amplicons, why are you seeing them? If the amount of template you're using is sufficient to produce fragment bands that bright on an EtBr-stained gel, you're using way too much template, and your PCR reaction would likely fail, even with perfect primers...
gyma on Apr 1 2010, 08:06 AM said:

Exactly why we must first investigate the source of all that banding...
Well, if those bands are not amplified by PCR, then why increasing cylce number resulted in increased amount? I attached a photo showing cycle number 30,35,40 from left to right in 1 PCR reaction of 1 carrier. I think you could see the brightness of all 4 bands increases as the cycle number increases. I am sure I didnt use too much template, 50 ng isnt too much for a PCR reaction, right?
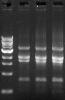
phage434 on Apr 2 2010, 03:20 AM said:
Your uncycled lane should show no bands whatsoever, because it should have sufficiently low template concentration that it is not visible.
do you mean the bands i got from PCR already exist in the template?
We're trying to figure out if those bands are amplicons. phage434's suggestion is designed to run two identical samples -- each comprising one-half of a PCR reaction mix that was made with twice the volume of everything you usually use in a mix, and then split -- thus the only difference between them is that one mixture was subjected to PCR cycling, and the other wasn't. If the bands are present and visible on a gel in the sample that wasn't cycled, they're not amplicons.
The whole thing comes down to the fact that your primer sequences were not in the insert you sequenced, meaning that that particular insert was not the result of a PCR amplification, and thus was a genomic fragment. What we really need is an answer to the question I posed way back here -- did you just sequence one or a few clones from a single transformation?
If so, you may have just accidentally recovered a genomic fragment of the appropriate size that was mixed in with a population of amplicons of the same size. Since you're blunt-end cloning, any fragment in a population of same-sized fragments has an equal chance of being cloned, regardless of whether it was generated by PCR or it was just a random genomic fragment that migrated with the amplicons.
HomeBrew on Apr 2 2010, 01:10 PM said:
The whole thing comes down to the fact that your primer sequences were not in the insert you sequenced, meaning that that particular insert was not the result of a PCR amplification, and thus was a genomic fragment. What we really need is an answer to the question I posed way back here -- did you just sequence one or a few clones from a single transformation?
If so, you may have just accidentally recovered a genomic fragment of the appropriate size that was mixed in with a population of amplicons of the same size. Since you're blunt-end cloning, any fragment in a population of same-sized fragments has an equal chance of being cloned, regardless of whether it was generated by PCR or it was just a random genomic fragment that migrated with the amplicons.
I just finished the PCR with a normal control from PBMC of a healthy person. although the normal control showed similar band patterns, it is quite different in the 2nd band, which is cloned and sequenced. if the 2nd band is specific, why does it contain human genome sequence?
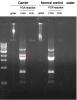
gyma on Apr 2 2010, 04:39 AM said:
HomeBrew on Apr 2 2010, 12:10 AM said:

HomeBrew on Apr 2 2010, 07:12 PM said:
gyma on Apr 2 2010, 04:39 AM said:
HomeBrew on Apr 2 2010, 12:10 AM said:
I sequenced 20 clones and they all have the same sequence, which proved to be a 2kb part of chromosome 13q.
From a single experiment? Could all the sequenced clones have been siblings? Have you repeated the experiment and also recovered the same 2kb part of chromosome 13q?
Here's the point -- if your 2kb part of chromosome 13q inserts were amplicons, then they would have the primer sequences in them. Since they do not, and it looks like the banding pattern you're seeing is a result of amplification based on your latest photo, we have to assume that the band your seeing is a mixture of amplicons (in vast excess) and co-migrating genomic fragments of the same size. During this experimet, just due to chance, you caught a genomic fragment when cloning, and what you sequenced were all siblings.
Strange things happen when cloning -- I once recovered a segment 100% identical to a tomato gene while cloning a band from E. coli -- no idea how it happened, but I repeated the experiment, and got the segment I wanted.
There are two major issues here -- your primers are not specific enough for what you are trying to do, as they produce way too many bands, and you're relying on blunt-end cloning. Primer specificity could be improved by re-design or changing the PCR conditions and/or cycling parameters. Blunt-end cloning could be avoided by attaching restriction enzyme sites to the ends of your primers, or, in this case, perhaps a better solution would be to try TA cloning. Use a Hi-Fi Taq-based enzyme, and clone into a TA TOPO vector. Either of these methods will insure that you can only successfully clone PCR products, and take the genomic fragments out of the equation.
HomeBrew on Apr 3 2010, 01:23 AM said:
I had ever successfully sequenced 2 carriers before using the same method, in that case, I got over 15 clones with the correct insert. I didnt care about the rest clones at that time. after this problem happened, I checked the sequences of rest failed clones, and I found the same sequence as what I got lately. So this might not be an accidental thing.
Here's the point -- if your 2kb part of chromosome 13q inserts were amplicons, then they would have the primer sequences in them. Since they do not, and it looks like the banding pattern you're seeing is a result of amplification based on your latest photo, we have to assume that the band your seeing is a mixture of amplicons (in vast excess) and co-migrating genomic fragments of the same size. During this experimet, just due to chance, you caught a genomic fragment when cloning, and what you sequenced were all siblings.
Strange things happen when cloning -- I once recovered a segment 100% identical to a tomato gene while cloning a band from E. coli -- no idea how it happened, but I repeated the experiment, and got the segment I wanted.
There are two major issues here -- your primers are not specific enough for what you are trying to do, as they produce way too many bands, and you're relying on blunt-end cloning. Primer specificity could be improved by re-design or changing the PCR conditions and/or cycling parameters. Blunt-end cloning could be avoided by attaching restriction enzyme sites to the ends of your primers, or, in this case, perhaps a better solution would be to try TA cloning. Use a Hi-Fi Taq-based enzyme, and clone into a TA TOPO vector. Either of these methods will insure that you can only successfully clone PCR products, and take the genomic fragments out of the equation.
Anyway, I should consider the next step, maybe TA cloning. I used Herculase II before, which is 6 times higher than taq in fidelity. I searched Hi-Fi taq-based enzyme and found easy-A from stratagene, which is also 6 times more faithful than taq. that is a choice. Do you have any recommendations?
Thank you very much, Homebrew. I really appreciate your help and I have learned a lot from you. wish i could solve this problem.
gyma on Apr 3 2010, 06:25 AM said:
We use the Platinum PCR SuperMix High Fidelity from Invitrogen. It too has 6X fidelity versus Taq alone.
There is another experiment I'd try. Prepare four PCR reactions -- one with both primers, one with just the forward primer, one with just the reverse primer, and one with neither primer added -- and cycle them all. See what banding pattern is produced in each case.
What are your PCR cycling conditions? Can you post your primer sequences?