| |
Top : Molecular Biology : PCR : Real-Time PCR : Real-Time PCR Quantification Using Cloned Standards and Multiple Housekeeping Genes
Real-Time PCR Quantification Using Cloned Standards and Multiple Housekeeping Genes | |
| Author: Wan-Jung A. Tsai, Diane J. Letina, Gayle A. Brazea | |
| Affiliation: Department of Pharmaceutical Sciences School of Pharmacy and Pharmaceutical Sciences, 517 Hochstetter Hall, University at Buffalo, Amherst, New York 14260, USA * Whom readers should direct their requests for reprints and other inquiries Director, Pharmaceutical Genetics Laboratory School of Pharmacy and Pharmaceutical Sciences University at Buffalo E-mail: dbrazeau@buffalo.edu | |
| Date Added: Mon Feb 02 2009 | |
| Date Modified: Mon Feb 02 2009 | |
| Abstract: We reported a general laboratory procedure for QRT-PCR, which addresses most of the inherent difficulties with this technique, can be easily adopted by any laboratory. | |
Procedures
Quantitative real time PCR (QRT-PCR) has become one of the most widely used methods of assessing gene expression owing to its sensitivity and reduced run time compared with traditional methods such as RNase protection assays, tissue (Northern) hybridizations, and the reverse transcription competitive PCR (1, 2). There exist several variants of the QRT-PCR procedure; each has its own advantages and limitations. A general laboratory procedure for QRT-PCR, which addresses most of the inherent difficulties with this technique, is implemented in our facility and can be easily adopted by any laboratory. This procedure is based upon SYBR green detection, though many of the quantification algorithms can be used with fluorescent probe technologies.
There are two methods of measuring gene expression using QRT-PCR: absolute and relative quantification. One of the most commonly used quantification methods in QRT-PCR is the delta/delta Ct method, which is a relative quantification method (1). The advantages of using this method are 1) elimination of the need for RNA or cDNA standard curves since the data is normalized against a calibrator, and 2) maximizes the number of samples can be run on one plate of an experimental run. The key assumption for using this relative method is that the amplification efficiency between samples and a reference (calibrator) are equivalent. However, amplification efficiencies, if calculated at all, have been shown to vary among the samples and experimental runs. As such, the real fold change comparisons cannot be made without introducing additional error. Our approach is to incorporate a cDNA standard curve in each PCR run and obtain an estimate of absolute copy number for each sample. Since the copy number is calculated based on the standard curve only, the absolute method is more robust against possible variations in amplification efficiency since efficiencies are calculated for every run. The cDNA standards are obtained by cloning the target sequence. Typically this is accomplished during the period when the PCR conditions are being optimized for each target. Cloning the target sequence using TOPO TA cloning kit is cost effective, convenient, fast (two days) and can easily be done in any laboratory setting. The plasmid inserted with the target sequence can be readily used as standards for the QRT-PCR.
With increased sensitivity to the target expression, the QRT-PCR also inherits high sensitivity to slight differences in the starting RNA quality and quantity, differences in the RT efficiency and PCR amplification efficiency between samples and runs, operator errors, and the presence of PCR inhibitors (3). This will inevitably introduce noise in the experimental data. Therefore, there have been many attempts to control these errors including normalizing the data to total RNA, 18s or 28s rRNA, or a single internal reference (housekeeping) gene. Each approach has its limitations. The total RNA method, while controlling the starting material, does not take into consideration the variability in the various steps in QRT-PCR. The 18s or 28s rRNA method overlooks that rRNA and mRNA are synthesized by different polymerases and therefore, the total mRNA amount extracted may not be best represented by the rRNA amount in the samples. The most widely used method is the single housekeeping gene normalization. It works under the key assumption the reference gene expression remains constant among experimental conditions. Therefore, the difference in the reference gene expression between samples truly represents the differences introduced from the starting material and the various steps in QRT-PCR. Recently, it has become clear that housekeeping gene expression fluctuates between samples or changes in response to treatments and as such, its suitability to serve as an internal reference should be validated for each experiment (4, 5).
Using housekeeping genes as an internal reference without proper validation of its presumed stability can lead to increased data variability, or erroneous results. Therefore, for data normalization, we routinely determine the expression of at least 3 internal housekeeping genes. A gene stability measure (M) by Vandesompele et al (6) has been developed to evaluate the stability of a given control gene by calculating the pair-wise variation with all other control genes as the standard deviation of the logarithmically transformed expression ratios. Based on the M value of each housekeeping gene, a normalization method was chosen for the experiment. One single housekeeping gene should be used if it has the lowest M value, or a geometric mean of the 3 housekeeping genes will be calculated as proposed by Vandesompele (6).
Serving as a second stringent control for the QRT-PCR, each RNA sample was spiked with a known amount of “alien” RNA (Alien QRT-PCR Inhibitor Alert, Strategene, La Jolla, CA) before being subjected to the reverse transcription reaction. Then along with the target genes to be assayed, the housekeeping genes, an aliquot of the reverse transcriptized cDNA was used to amplify the alien gene using the supplied primers following the manufacturer’s protocol. This step allows us to accurately monitor potential inhibitors in PCR conditions.
One potential draw back to this approach is differential efficiency between the standards and the true samples. In order to calculate the efficiency in the samples, we performed an experiment in which serial dilutions of the samples and the standards were included in the same real time PCR run. Amplification efficiencies were similar between samples and the standard and thus supported the use of a standard curve as a more accurate quantification method.
This general Q-RTPCR protocol is summarized in Figure 1 and it can be easily adopted by any laboratory. It is a robust quantitative method for gene expression studies. We highly recommend constructing a standard curve for the gene of interest because it offers an accurate measurement of the mRNA without assuming equal efficiencies between the samples and the reference and the ease of obtaining standards by cloning the amplicon using a commercially available kit. Multiple housekeeping genes should be validated for normalization in each experiment and incorporation of an external standard (alien RNA) is also needed to identify possible PCR inhibitors between treatments and runs.
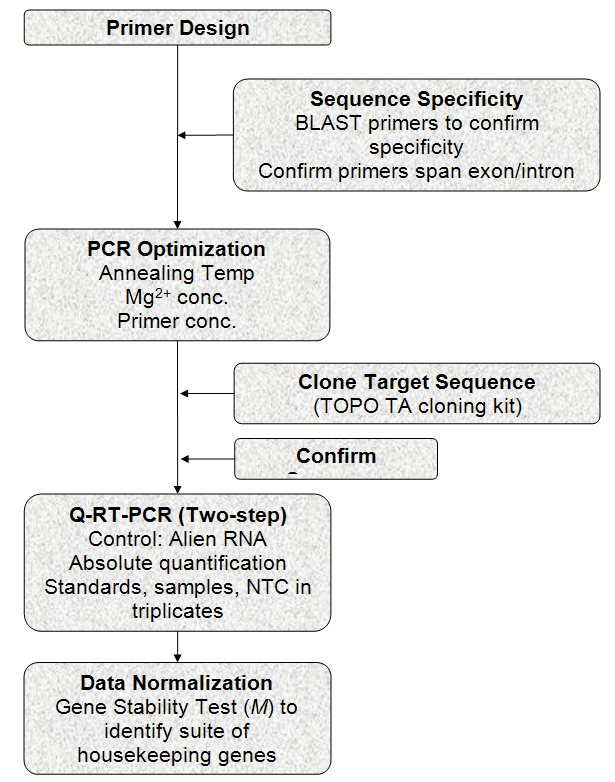
Figure 1. General laboratory procedure for Q-RTPCR using cloned standards and multiple housekeeping genes.
Acknowledgements
This work was partially supported by Kapoor Foundation funds, School of Pharmacy
and Pharmaceutical Sciences, University at Buffalo.
Competing Interests Statement
The authors declare no competing interests.
References
- Bustin SA. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol 25:169-93, 2000.
- Wong ML, Medrano JF. Real-time PCR for mRNA quantitation. Biotechniques 39:75-85, 2005.
- Bustin SA. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol 29:23-39, 2002.
- Suzuki T, Higgins PJ, Crawford DR. Control selection for RNA quantitation. Biotechniques 29:332-7, 2000.
- Thellin O, Zorzi W, Lakaye B, De Borman B, Coumans B, Hennen G, Grisar T, Igout A, Heinen E. Housekeeping genes as internal standards: use and limits. J Biotechnol 75:291-5, 1999.
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:RESEARCH0034, 2002.