Primer dimer issue in real time PCR - (Jun/22/2009 )
hello every one
I am very new in real time PCR with SYBR green. I am Checking 2 gene expression anog with endogenous control. In meling curve I am getting an additional peak for 1 gene( in NTC also have the additional peak)but other 2 showed only one preak. but when I increase the template amout the additional peak dis appeared. but I dont know can I use different amount of template for different gene while using delta delta CT method
(relative quantification)--May I?
hope get suggestion
panna
Hi,
if you want to detect and quanify SNP without high-resolution melting, it is also possible. You have to design primers with one extra mismatch at the penultimate position (second to last of the 3'-end). See https://www.biomedcentral.com/1471-2164/8/275
This is their primer design tool which wil help you do the job: https://bioinfo.biotec.or.th/WASP/index/help
I designed the primers that way and I am able to detect and precisely quantify two variants of SNP in template mixture very well using just SybrGreen in Stratagene Mx3005 machine. I am running two reactions - one for each SNP variant. Then check the melting curve and compare the Ct.
But you have to test it first on control samples - for example mixtures 1:1, 1:10, 10:1 etc. of SNP variants.
It is cheap SNP quanification method for those who do not want or cannot spend money on Taqman probes.
Thanks everyone for explaining the NTC and primer dimer in real-time pcr. 
The HRM mode should be used to distinuish one base pair mismatch between two templates
Template-dependent primer-dimers
I just had an idea, maybe you already thought about this or even wrote about it somewhere in the forum, but this striked me just now, so I'm writing it here because it's a sticky topic and someone may be interested. (or maybe as said in czech, I just discovered America - something already well known). 
While using SYBR method (mainly UPL-designed primers that eventualy were used without the probe) I quite often encountered template-dependent primer-dimers, NTC was positive for a product with lower melting temperature, probably dimers, but there were no dimers in any sample, just the specific peak. Since it wasn't interacting with sample measurements I just let it go, but I always wondered, why there are dimers in NTC but not in samples, when there all the same except template?
Just now i realised what actually is different in NTC, the amount of free Mg+ ions!
DNA captures Mg+ ions (thanks to someone who mentioned it somewhere) and decreases the free pool of them. But in NTC there is no DNA and there are more Mg+ that makes the mix less stringent. In less stringent enviroment, the dimers form but they don't in samples with a higher stringency.
This could be the perfect reason why there are template-dependent dimers. What do you think, makes sense?
I ve gone through the entire qpcr forum and I can not find answer to my problem. Please help its driving me crazy, sleepless nights etc etc and I am doubting my competence. Ok this is the story:
I have 4 groups of mice with 9 mice per group. Ive extracted RNA using Qiagen kit for mRNA extraction including miR (including DNase treatment). Checked all my samples on nanodrop, and quite a few had an extra peak. So I cleaned them up with 5M Sodiium acetate method. Finally beautiful looking samples when I nano dropped them. To make sure I m really using the 400ng for my RT , I double measured all my RNA samples with Qubit assay (Invitrogen). Did RT with promega kit, random hexamers.
I made std curve by pooling 20 samples and then 1/3 serial dilution. All the time using RNAase free water. And I also diluted my samples 1:50 and 1:20 (falls into my std curve). (PS I ve run 2 of the samples on RNA seq so I m now in the process of validating the results by qpcr, already done PCR and seen that the primers work).
I ve run 5 different housekeeping genes (2 were ABI probes and 3 were sybergreener) on the std:s and the curves come up nicely. So I went ahead with my samples. My triplicates are very nice both for the housekeeping and gene of interest. But I get five different patterns on the same samples /dilution dependent on my housekeeping. QUESTION 1, what is the right pattern, what to trust? Then I looked back at my housekeeping and found that the ct values are sometimes 5ct values difference between samples while my gene has only 1-2 ct difference. And that on a GAPDH probe. I looked at the others and same problem there. Can I really have 5 different housekeeping genes , all regulated somehow? What shall I do?
Because my boss is stressing me out wanting results, I have just continued hoping it was just for that gene. So I ran my stds on all 18 genes of interest (10 syber and 8 probes) and IT ALL LOOKS HORRIBLE. I have primer dimers i my sybergreener, some are present only in the lower dilutions, and some in the higher dilutions of the std curve. How do I solve that? My Tm is around 68 and I am acquiring at 60c.
I am stuck. And my boss has threatened to give my qpcr tasks to someone else in the group because I cant produce what he wants to see. HELP
First, you have problems with stable housekeepings, you have to search the literature for housekeepings used in experiments silmilar to yours to pick out those that should be stable (GAPDH even when used widely is not stable at all conditions, like hypoxia experiments, in fact many housekeeping may be involved in the same pathways) and use geNorm or ...(what was the other?) to select the most stable one(s) in your setting. You may try ribosomal proteins and so on. For miRs you usually need miR reference, there are some published.
You seem to dilute your samples pretty much, but that's dependent on the abundance and RT. If you get Ct between 15-30 it's probably OK.
As for the dimers, you need to optimise the reaction, increase temperature, change Mg. Do you get dimers together with your product or sometimes only in negative samples (including NTC)?
Hey everybody,
this is my first post in this forum and I wanna just add my qPCR problem to this thread:
I am not really sure, what went wrong in my primer efficiency tests, but I am assuming that I got primer dimers (small shoulder??). But additionally to this problem, I have peaks in my no template controls. What can this be? I loaded a gel and I could not see any bands in my NTC-lanes, pointing to any unspecific amplification / contamination.
Please see the attached file.
best,
sebastian

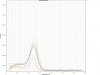

What are your "standards"? They didn't look right, the peaks are wide and short.