Problems with my ligation reaction - (May/20/2009 )
Hi there,
I am a complete newcomer to molecular cloning, and I am having some trouble ligating a 1.7Kb insert into a 4.8Kb vector.
To generate my insert I PCRed sticky ends onto a cDNA that I had in a different vector ( the ends contain sbfI and hindIII sites 5' and 3' respectively), then I gel purified this product using the QIAEX II kit and made sure that this is indeed the insert I want by digesting it with different restriction enzymes. Then I double digested with sbfI and hindIII in NEB buffer 4 ( I know the HindIII has 50% activity in this buffer) and purified again.
For my Vector (Initially 5.1Kb) I double digested 2ug overnight with sbfI and hindIII in NEB buffer 4, ran it out on a gel and extracted a band at 4.8 Kb. I made sure that I saw the 300bp fragment by doing the extraction on a 1.5% gel. ( I extract and purify with the QIAEX II kit)
So now I have the purified, digested vector and the insert and I have been trying to ligate them together with both the quick ligation kit and the regular T4 ligase. I use a 3X the vector rule for adding insert into both reactions, and the total DNA concentration is probably 200ng (vector + insert). I haven't been adding extra PEG to the slow ligation reaction, but I add the DNAs the ligase and the 10x buffer into 10ul total volume and incubate in an ice bucket at 16 degrees overnight ( so the ice melts and the temp slowly climbs to RT). I have also tried this with CIP and no CIP.
My quick ligation reactions do not seem to be as successful, but after reading a bit online it looks like the insert and the vector may not have enough time to anneal if the reaction is only run for 10mins.
Anyway, the problem is I either get no colonies OR I get way too many, none of them having incorporated the insert.
After reading these forums and the archives it looks like ligation can be troublesome. Is there some newbie mistake I am making?
Any suggestions would be much appreciated
Arjun123 on May 20 2009, 12:49 PM said:
Did you add bases to the ends of the restriction enzyme recognition sequence (5' bases for the 5' cut site, and 3' bases for the 3' cut site) to ensure that the RE has enough DNA to be able to bind?
bob1 on May 21 2009, 11:15 AM said:
Arjun123 on May 20 2009, 12:49 PM said:
Did you add bases to the ends of the restriction enzyme recognition sequence (5' bases for the 5' cut site, and 3' bases for the 3' cut site) to ensure that the RE has enough DNA to be able to bind?
Once you have checked out that piece of advice, and corrected if necessary, I would also consider doing sequential digestions. 1 or 2 extra hours is a lot less hassle than a repeated experiment. Go to the NEB website for their advice on doing sequential digests.
Once you have done your digestion, try a quick RT ligation on some of it, then run a gel. You should have a concatamer of the insert; if you only get a dimer-sized product, one of the sites hasn't cut well. Test the ligase itself by treating some DnA ladder.
Yeah, I have 5bp on the 5' end (past the sbf1 cut site) and 6bp on the 3' end (past the hindIII cut site).
But that's great advice! - so If I just add the PCRed and digested insert alone into a room temp ligation with the NEB t4 ligase that I use along with an undigested control then I should get some kind of ligated form of insert-to-insert? ( I am guessing this is what a concatamer is - I'll read up on this more)
I'll also try the ladder ligation as well.
I was wondering if there was a way to test whether the insert is cut well - I'll try this out right away and let you guys know
In our experience, heat killing the ligase prior to running the gel improves things. I recommend against doing this prior to transformation.
hi there
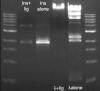
So I have did a ligation with my PCRed insert alone and with lambda DNA. Lambda DNA ligates nicely. But my insert only shows one ligation product. The insert is 1.7 Kb and I see a band at about 4Kb. The double of 1.7 should be about 3.4, but I will run the gel out longer to get a better Idea of what is happening.
Is this what a concatamer should look like? I tried to look around for pictures of gels on the web of what I should expect but I couldn't see anything. Should I only expect one band?
I've attached a picture of my gel:
ins+lig: 4ul insert (100ng), 1ul 10x Buffer, 1ul NEB T4 ligase, 4ul RNAse free H20.
ins alone: same as above, no ligase
lambda+lig: same as above but with 1ul of lambda DNA
lambdaalone: same as +lig but no ligase
The ladder is an NEB 1KB ladder, from bottom to top 500bp 1Kb 1.5Kb 2Kb 3Kb 4,5,6,8,10Kb
What do you guys think?