RT-PCR primer design guide - How to check gene structure and design the primer? - Recovered post (Jan/29/2009 )
RT-PCR primer design guide - How to check gene structure and design the primer? <small> (Nov/03/2003 )</small>
I would like to ask generally how you design your RT-PCR priemrs and specifically, how you check gene structure to contain some introns in the amplified region. Please let me know where is the best place to get such information.
thank you.
ZW
-zhongw90-<hr>
Hi ZW,
A potential problem with RT-PCR is DNA contamination in RNA. To tell whether amplication is from cDNA template or genomic DNA, RT-PCR primers are usually designed to span introns or bridge an exon-exon junction. When two primers span one or more introns (Fig.1), the amplified product from genomic DNA will be bigger than expected. Primers can also bridge an exon-exon junction (Fig. 2). Such designed primers will not amplify genomic DNA template because the intron will intervene the pairing between the primer and DNA. But, make sure that only a few nucleotides of the 3' portion should pair to the 3' exon as shown in Fig. 2. If the 3' portion of the primer has substantial pairing with the 3' exon, it can still initiate amplication without its 5' portion pairs to the 5' exon (Fig 3. sense primer).
This is how I usually design RT-PCR primers:
I. First get the cDNA sequence of my gene from NCBI. I only use Refseq if available. RefSeq represents the most reliable and unique cDNA sequence for a gene. Because searching the nucleotide database is not effective (you may get hundreds of sequences for a gene, most of them are irrelevant) so I will go to Entrez Gene database (previously LocusLink) at https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene to find the gene of my interest, then scroll down to the NCBI Reference Sequences (RefSeq) section to find the RefSeq mRNA sequence whose accession number usually looks like NM_xxxxxxx. Then, click the sequence link which will lead to the actual sequence from GenBank Nucleotide sequence database.
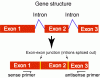
Figure 1
![]()
Figure 2
![]()
Figure 3
2. Copy the sequence to a program to design primers such as Primer3 at https://frodo.wi.mit.edu/primer3/. Just accept all default parameters provided by the program and design primers. Choose the pair with highest score.
3. Copy the sequence corresponding to the amplified region by the primer pair you have chosen and then go to UCSC genome browser at https://genome.ucsc.edu/cgi-bin/hgBlat and paste the sequence there and do a BLAT.
4. Check if the amplicon spans any introns by view the detailed alignment of the query sequence against genomic sequence.
5. Or do a BLAT using the whole cDNA sequence first to know where exon-exon junctions (on cDNA) are and then design primers by specifying a target region corresponding to the junctions
6. Another purpose of Blatting amplicon sequence against genome database is to check if the amplified fragment has any homology with other genomic regions. The Blat results will tell that.
7. There are some other places you can also check gene structure such as NCBI Aceview at https://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/index.html and Ensembl at https://www.ensembl.org,
Edit: Sept. 6.
UCSC also provides an in silico PCR tool which allows you to input a pair of primers and retuns potential amplicon targets in the genome. It is useful for checking off-target amplification by your Rt-PCR primers. You can find the tool here: https://genome.ucsc.edu/cgi-bin/hgPcr
Hope that is of some help
-sage-<hr>
Agree.
I have a question:
Which part of a cDNA sequence (5' UTR, 3' UTR, ORF, or any motif region) will you choose as amplifying target for RT-PCR, if they satisfy all the requirements (uniqe in genome, average GC, etc)? My feeling is they should all be OK for expression study. Am I right?
Kawaka
-kawaka-<hr>
<!--quoteo-->
QUOTE <!--quotec-->Agree.
I have a question:
Which part of a cDNA sequence (5' UTR, 3' UTR, ORF, or any motif region) will you choose as amplifying target for RT-PCR, if they satisfy all the requirements (uniqe in genome, average GC, etc)? My feeling is they should all be OK for expression study. Am I right?
Kawaka<!--QuoteEnd--><!--QuoteEEnd-->
Is it right???
-lyrezxl-<hr>
Hi All !
Notice that to study gene expression, the best is to design primers on two different exons, thus you obtain something specific for mRNA/cDNA, because you can distinguish cDNA from genomic DNA
Take a look here... this could help !
https://www.biocompare.com/techart.asp?id=799
-freb-<hr>
Hi ZW,
first a warning: I'm from the company making this software.
But you could try www.probelibrary.com/adc which designs intron-spanning real time PCR assays on the fly. Only, you have to use Probelibrary Probes for these assays.
I would be interested in knowing your opinion of the tool...)
Søren
Søren M. Echwald, MSc., Ph.D.
-----------------------------
Exiqon A/S
Bygstubben 9
DK-2950 Vedbaek
Denmark
www.ProbeLibrary.com
One Real-time PCR kit, which covers 38.565 genes
-smesme-<hr>
Kawaka wrote:
<!--quoteo-->
QUOTE <!--quotec-->Which part of a cDNA sequence (5' UTR, 3' UTR, ORF, or any motif region) will you choose as amplifying target for RT-PCR, if they satisfy all the requirements (uniqe in genome, average GC, etc)? My feeling is they should all be OK for expression study. Am I right?<!--QuoteEnd--><!--QuoteEEnd-->
I've had some problems with RT-PCR when using primers in the 5'UTR region. A gene may have different transcription initiation sites in different tissues, maybe even in a same cell line under different conditions. As an example, I was not able to amplify a gene from total pancreas RNA using the upper primer in the 5'UTR (nucleotides 69-80 of the RefSeq sequence) but the same primers worked wonderfully on liver RNA. I changed the primers to the exon2-exon3 region and then I got similar levels of amplification from liver and pancreas. It is not a case of alternate promoters, the exon 1 is the same in both tissues, it's only that different transcription start sites a few nucleotides upstream or downstream may be used preferentially in the two tissues.
Hope this helps. Cheers.
-badcell-<hr>
<!--QuoteBegin-kawaka+Jan 23 2004, 11:53 AM-->
QUOTE (kawaka @ Jan 23 2004, 11:53 AM)<!--QuoteEBegin-->Agree.
I have a question:
Which part of a cDNA sequence (5' UTR, 3' UTR, ORF, or any motif region) will you choose as amplifying target for RT-PCR, if they satisfy all the requirements (uniqe in genome, average GC, etc)? My feeling is they should all be OK for expression study. Am I right?
<!--QuoteEnd--><!--QuoteEEnd-->
If you use oligo dT primer for RT reaction, it is advised to design RT-PCR primers half way to the 3' end of the mRNA sequence in case that the RT reaction for some genes doesn't extend long enough to the 5' portion.
-pcrman-<hr>
It's everything very nice. I have one question though, probably simple. If my gene has pseudogenes, I mean with less or no introns, where do I look for their sequence, to design primers that do not amplify pseudogenes? However hard I can try to get rid of DNA, there's always a possibility that there's something left and the pseudogene will amplify instead, especially if it's a low-copy gene and I put a lot of cycles.
-Telomerase-<hr>
<!--quoteo(post=7404:date=Sep 23 2004, 02:41 AM:name=smesme)-->
QUOTE (smesme @ Sep 23 2004, 02:41 AM) <!--quotec-->Hi ZW,
...<!--QuoteEnd--><!--QuoteEEnd-->
You can also try AutoPrime at www.autoprime.de.
It does the same thing but gives you the option to change the parameters, choose different output formats, etc.
Greetings,
Flexer.
-flexer-
Hello everybody!
Am a new member of this forum, am a research student.Can any one of you help me regarding this.I have a gene sequence of lacZ from E coli .I had designed primers for its amplification and it worked now I want to design RTPCR primers and will use total RNA from E coli as template and want to maake cDNA first so how do I design RTPCR primers so that from the cDNA I can later amplify the lacZ gene???Please help me out
-codon-<hr>
Probe library from Exiqon or Roche-applied biosciece is good in finding primers which span an intron
-srisashi-<hr>
Here is the SOP in our lab, I got an almost 100% (ot of 30 genes I tried) accuracy for rt-pcr. However, there is something like 60-70% success in QRT_PCR without trying any optimization in PCR parameters or concentrations...
Designing Primers for Real Time PCR:
PERL PRIMER is the program of choice...
Download it from: (click this to see documentation..)
https://perlprimer.sourceforge.net/
In realtime PCR primer design, following points should be considered...
1. Primers should encompass an intron to see any genomic contamination.
2. One of the primers should reside on the intron exon boundry, (most of the primer length should reside on the 5' exon to prevent mispriming...)
3. A GC clamp should be present on the 3' most end of the primer to prevent mispriming.
4. The amplicon size is recomended to be between 100-300 bp.
Primer Design Protocole
1. Go to the Entrez Gene database.
2. Find the REFSEQ of your gene.
3. Get the refseq cDNA of your gene...
* There is a link to the REFSEQ db from the ENTREZ GENE entry of the gene...something like NM_XXX....
* Click FASTA format sequence of the REFSEQ cDNA,
* Save the line starting with a greater than ( > ) sign as the identifier of your sequence...
4. Go to BLAT ( https://genome.ucsc.edu/cgi-bin/hgBlat )
5. Get the genomic sequence which corresponds to your gene by pasting your cDNA sequence in to BLAT and clicking submit.
6. Then just click find primers on perl primer, after pasting the genomic and cDNA sequences of the gene.
7. Finally, after you select your primers check the products which might be produced by these primers by epcr tool of NCBI (https://www.ncbi.nlm....pcr/reverse.cgi). Read the help of that tool if it is first time...
* Enter your primers in 4 column format in REVERSE STS search tool: Four-column format, exactly one STS per line, columns are: label for sts, left primer, right primer, and product size (can be two dash-separated numbers for range of sizes).
!!Record primer information with at least the following information in an excel file
* Primer name (Gene symbol, primer number(should be unique in the lab), F or R, e.g: ACTB1F, ACTB2R)
* Target gene
* Primer sequence
* TM of primer
* Unique accession number of the sequence from which primer is designed. (gi number of NCBI is a good option, do not use gene accession numbers or gene symbols, they do not identify a sequence, sequence information for genes are always changing!!!)
* Length of the primer
-ersenkavak-<hr>
I got so much from this discussion. thank you all here. But I have a question, has anyone investigated how seriously (to what extent) the NDA contamination affects the RT-PCR results?
-rickyama-<hr>
let me add my own opinion about the DNA contamination in RT-PCR.
i am doing Ratio-RT-PCR to investigate the expression of N-cadherin in cardiomyocytes.
this method is to express the mRNA level by presenting its ratio to some reference. here for example, if i have 100 cells, then the gene in DNA of the target should be 200, let 0.1 be the contamination rate (it is not so low). then the DNA contamination in cDNA should be 20. then let estimate the number of cDNA. in a cell, one gene expresses 10 mRNA (is it reasonable?) then we will roughly have 1000 cDNA, so the contamination rate before PCR should be 2%. I do not think this data constitute a big problem for Ratio-RT-PCR. therefore, spanning different exons is not so important for the primer design in Ratio-RT-PCR. is anyone agree with me? or any comments to the above analysis? thank you.
-rickyama-<hr>
Primer3 plus is one of the most comprehensive primer design software available. It will remove the guessswork involved in primer design. You can also look for rt-pcr primer design tools such as PCR Now -(https://pathogene.swmed.edu/rt_primer/), the pcr suite and qPCR primer design tool -Beacon designer.
-AshRo-
Hi sage,
i found your information is really helpful. thank you so much 
hi all,
I know this may sounds so wrong, but if any1 out there having this oligotech software to spare me? I need it urgently. Thanks.
Please do find me via cheerioet.sp
thanks in advance.
Cheerioet
Hi. I'm designing RT-PCR primers, and i just got as far as step 4.
I entered the region between the two primers into the BLAT search. My results looked like this:
ACTIONS QUERY SCORE START END QSIZE IDENTITY CHRO STRAND START END SPAN
---------------------------------------------------------------------------------------------------
browser details YourSeq 194 1 196 196 100.0% 4 + 169589442 169604199 14758
browser details YourSeq 21 94 114 196 100.0% 2 - 103130734 103130754 21
browser details YourSeq 21 116 136 196 100.0% 9 + 86016389 86016409 21
browser details YourSeq 20 136 155 196 100.0% 5 - 76441233 76441252 20
browser details YourSeq 20 153 174 196 95.5% 1 - 38465325 38465346 22
browser details YourSeq 20 153 172 196 100.0% 20 + 21567108 21567127 20
However, i do not understand what to do next with this. Can anyone help?
Now NCBI provide a very convenient tool called Primer-BLAST for RT-PCR primer design. You can just provide a sequence or sequence ID, and specify whether you want the primers to span exon-exon junctions or include introns, the program will take gene structure into consideration.