Plasmid analysis in limited conditions - (May/02/2018 )
We use mammalian expression plasmids that show repeated (but inconsistently) problems as failure to transfect (depending on transfection reagent, FuGENE or PEI), uneven transfection pattern on WB, gelly consistence after several months, but even as one month old. Common solution is just to make new ones (or try to boil them for 10 minutes, but allegedly that doesn't help that much), but the problems keep happening once in several months to different people, I suspect there to be a systemic problem.
I would normally start at the beginning and try to check everything, but due to certain resons I'm not allowed to. Also, there are several things done not in plasmid-GLP way, some of which I tryied to avoid, but others I can't.
Plasmids are isolated from E.coli. on a Invitrogen HiPure column, but it is overloaded (recommended volume for high copy plasmid is 15-25 ml of culture and we use 100 ml). Nonetheless the yields are good and 260/280 purity as measured on Nanodrop is 1.9 (the 260/230 is also fine). And they seem to work, at least at the start, with storage concentration around 1 ug in low EDTA TE. Commonly they are disolved overnight at fridge and measured, diluted and stored in -20, thawed for each use (which could be like once a week). But mostly they would be overdried before that, as this is difficult to prevent with processing several plasmids at once.
What I did try was to carefully get rid of any ethanol wash remains, try not to overdry pelet and keep it constantly in 4 C (and before that 1 hour on 37 C), and so far they seem to work (with PEI). But also I needed to see if that has any effect.
I have limited means to test them, I had only a few REs to use, I can run uncut on gel, sequence with common plasmid primers and test different conditions of storage, but that's all. So I mainly focus on bacterial gDNA contamination, plasmid size and plasmid conformations.
My first (frozen-unfrozen, a bit gelly after a while) batch I run on gel and seen a huge bright wide blob in between where the plasmids and gDNA should be and visible signs of undissolved DNA (the bands with these rectengular-like tail edges). That and something about frequent freezing-thawning cycles possibly creating a small microenviroments around DNA, that could affect solubility, I decided to keep them in fridge further on, as they should be stable anyway for such short time we use them.
The I used a single cutter to check the size and they have the expected 8 kb, also fully cut they do not exhibit any smear or blob, so I concluded that the smear had to be plasmids (possibly some multimers??).
Anyway I never seen the blob since, while I keep them in fridge, so that should have been gone.
But now the very stupid question at the current end of my testing, when I run 100 ng on 0.8% agarose (0.5x TBE buffer) I see the most bright conformation to be much higher that 8 kb (and 5,5 kb for empty) even though the supercoiled should run even lower than that. My colleague told me, that that's the effect of EtBr binding and it is the supercoiled, but the same happens when I decrease the amount of EtBr to 1/2 and I'm still not sure, that it could cause such a shift and I didn not ever seen that before on plasmid checks.
I was wandering then if that means that the most abundant now is not the supercoiled variant. Or maybe not using TAE for plasmids (that I can change).
I'm aware that transformation efficiency varies with different conformation and used method, usually supercoiled are preferable. Any ideas welcome.
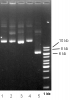
(lines 1-2, 8 kb plasmid, 3-4 something around that size too, 5 is 5.5kb empty backbone)
That is a problem.
Super-coiled can run very strangely, sometimes it will run larger than expected, sometimes smaller. The ethidium should not impede the migration that much. If you have access to a bioanalyzer you could try running your plasmids through that to verify. I would be surprised if you are getting almost 100% super-coiled, and that components would run that much bigger than expected. Could you try linearizing and see what happens?
Overloading the columns could be a problem - this will let some of the cytotoxic inhibitory components come through the prep and into the DNA, which could be causing some of the effects you are seeing. However this would depend on how much bacteria you are getting in your culture. If you are growing under ideal conditions for e.coli, then you will be definitely overloading.
Freeze/thaw cycles can affect plasmid integrity, but shouldn't alter the ability to transfect etc. If anything linearized plasmid should transfect better than circular.
Linearized are roughly the expected size (at least those 8 kb ones - 8 kb), for the others I was not sure about the backbone. Unfortunately Bioanalyzer is not available.
Thanks for the notes about transfecting. The cultures are overnight, well grown and the density I would say optimal. But unless I can prove this does have a serious effect on how long the plasmids can be used, sadly I have no argument to change it.
Meanwhile, I was able to sequence plasmids in lines 3,4 and 5 (empty) with a T7 fwd and BGH rev primers. Empty seems to be a pcDNA3 backbone, the one that Invitrogen updated to 3.1 due to a presence of hairpin in the MCS:
What is the difference between pcDNA3 and pcDNA3.1 vectors?
Answer:
pcDNA3 is no longer available from Thermo Fisher Scientific but has been directly replaced by pcDNA3.1, which was derived from pcDNA3. The center of the multiple cloning site (MCS) within the original pcDNA3 vector contained homology to a hairpin mRNA structure and involved the Eag I, Not I, and both BstXI sequences. This hairpin would only have affected expression of genes cloned downstream of the Not I site, if at all. To address this issue, some sequences were removed, including the Eag I site, and the BstXI sequences were slightly modified to reduce homology. A 32-base fragment from pcDNA3 (between bases 995 and 1026), which contains the Sp6 primer site, was also removed and 11 bases were inserted in its place, adding another PmeI restriction site into the MCS of pcDNA3.1.
Answer Id: E9155
Plasmid in lane 3 seem to be in the same backbone. Both had pretty bad fwd sequences and the sequencing company suggested presence of a hairpin. (plasmid 3 has an ORF cloned just downstream of NotI)
Plasmid on line 4 is likely a pRc/CMV and that also had a very bad fwd sequence (with T7 fwd).
All of then however had very nice readable rev sequences. I had no experience with plasmid secondary structure causing troubles, except for retroviral vectors that can recombine in E. Coli, but at least the plasmids I did send, had so much a hairpin, that they were difficult to sequence. The #4 seem to have a similar problem, but pRc/CMV should not have this kind of known drawback. Also, it is possible the T7 primer doesnt work well, they could all be sequenced with CMV fwd, which I will try.
But, if in any case, the plasmids do make hairpins and other weird structures (even because of the overloading) that could create shift in mobility. Or/and more disrupted plasmids could be more prone to this behavior, which could cause unpredictable outcomes after some time.
If you have T7 you might well have Sp6 in there as a candidate for sequencing.
I have never heard of hairpins being a problem for sequencing with most plasmids. I would be very suspicious that it is happening in all/most of your stocks. It sounds like you might be getting a lot of glycans or other cell wall components coming through the preps - these would read pretty similarly to DNA/RNA on the spec, and could cause aggregation of the DNA into gel-like masses. They probably wouldn't stain with EtBr, though I've never looked to see if they would. They would also interfere with PCR/sequencing. I've had this problem with plant DNA extractions before (nearly 20 years ago now...)
Any chance you could do a prep without overloading the column and see what happens?
The other cell type contamination could be a culprit, but I have no way to test it.
I did another prep with half the culture size and increase the lysis/neutralisation buffer volumes. But the problems happened after some time and unpredictably (some plasmids I use are several months old and transfect fine, others do not). My colleague did say he is doing it the same way, rather than the "common practice". So I will see.
Hairpin is a problem for sequencing with most plasmids? I found something about plasmid sequencing yesterday. If you are curious about this topic, here is the lik: Complete Plasmid DNA Sequencing
I have also noticed that stored plasmids, particularly large ones (over 10 kb) tend to slowly degrade over time; they become gelatinous and won’t go back into solution. This is particularly true if they are stored in high concentration (over 500 ng/ul) and have been freeze-thawed too much. I suspect some kind of permanent denaturation, though exactly why/how I don’t know. But sometimes I have seen them precipitate out and they will not redissolve. Using freshly-prepped plasmid may help with your transfections, and if that is too costly to do in a midi-prep format, maybe a fresh mini-prep will do. The only way to really assess whether your bacterial culture volume (overloading) is at fault is to do a controlled experiment comparing the same plasmid prepared according to the kit directions (25 ml) and overloaded (100 ml) and treat them exactly the same afterward (same storage conditions, concentration, etc.) Then use them in side-by-side transfection experiments at various time points after storage. Obviously this is costly and if you are not in a position to choose to do this, that is unfortunate. It would be an interesting experiment.
Another thought: What host cells are you using? Are they any of the old strains that produce nucleases?
And this is a real unlikely possibility, but check the pH of your TE, as I once found that an older bottle of commercially-sourced TE had gone “bad.” It had acidified. I don’t know how, but it was stored on a shelf near a sunny window and that possibly affected it. Plasmids I had resuspended in that TE went “bad” fairly quickly. It took me quite a while to figure that out!
I have also observed that I get better sequencing results with fresher plasmid preps, even when the older preps still cut correctly with REs. There is usually a gradual conversion of supercoil to open circle/nicked forms with time, however. Probably the nicked stuff was affecting the sequencing quality.
Trof, your original post was a while ago- did you solve your problem?