Restriction Digest with pGEMT for TA cloning - (Mar/27/2017 )
I have transformed my DNA using pGEMT vector in E. coli. I have Nde1 and Xho1 as my up and downstream overhangs. My question is: Do i have to do digestion using EcoR1 alone first, streak another plate and send that for sequencing to confirm my insert? Then, i can do double digest with Nde1 and Xho1?
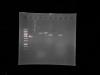
I went ahead and did double digestion without going the EcoR1 route. I have attached the results of the gel for that, were lane one is 1kb ladder, lane to is control without the enzymes, and the rest are my samples.
Can anyone help please???????
I'm a little lost here - why do you want to do an EcoRI digest, and why are you streaking plates?
Normally if you have an insert you will either be able to see a size shift compared to the empty vector (insert size dependent, of course); or you can do a simple screening like PCR amplification using one primer that is based in the insert, and another that is based in the plasmid.
For your gel, you need to tell us what we are seeing - how big is the plasmid to start with? How big would it be with insert? Is this just insert we are seeing or is it plasmid and insert? What are the controls, and what should they look like? How much DNA did you load?
EcoRI has restriction sites on both sides of the insertion point in pGEMT. Meaning if i expose my sample to EcoRI, i should end up with 2 sorts of fragments: 1 representing the vector (300bp or 3kb) and the other representing my insert (1197bp). The streaked plate is the one with PCR colony, sorry for that, which i have to send for sequencing to confirm my insert.
For the gel: The plasmid is 3000bp long, so with insert it should be 4197bp long.
The first lane is 1kb ladder, lane 2 is plasmid with insert but NO restriction enzymes (control), and the rest are my insert + Vector + enzymes. 6 samples in total but the first sample is the control. In each i added 3uL of my DNA, 42uL water, 5uL O buffer and 1uL of each restriction enzyme.
I do however, feel like my transformation could not have worked but what is those bands on the Gel representing? My insert can NOT be higher than the vector.
Ok, I understand now. As a screen I would do colony PCR directly off the colony - for this, I would pick a colony, streak a small line on a fresh plate (as a backup, and so you have something to go back to when you get a positive, use the same plate for lots of streaks, just draw a grid on the bottom so you know which is which) then dip into 100 uL ultrapure water (or the other way around; dip then streak). Use 1 ul of the water in a PCR with either primers anchored on either side of the insert, or primers for the insert only, or with a primer on the plasmid and a primer on the insert. For options 1 and 2 there you can have a positive control hopefully, the third option doesn't have a positive.
As you are putting a big insert into a relatively small plasmid, you should easily be able to see the size shift on a gel, as it will change from about 3000 to over 4000 bp. To do this, ideally you would linearize the plasmid so that you are not contending with supercoiled DNA and those sorts of things, so a single cut should be sufficient. Your EcoRI digest should be fine too, though you should include a positive control so that you know it is cutting. For digests you need to make sure that you are digesting the right amount of DNA under the right conditions - use an excess (2x) of enzyme relative to the DNA amount and do a short digest (1 h at 37 C is common), not overnight or any extended period.
From your gel, I would guess that your digest hasn't worked, but it is a little hard to tell as I can't work out the size of the bands. If you can work out the band sizes, then you should be able to tell what is going on. Are you sure this is 1 kb DNA ladder from Invitrogen/Thermo fisher, could it be 1 kb plus ladder or from another company? You also say you have a control of DNA + insert with no enzyme - which sample does this DNA correspond to, or is it an unrelated DNA that is cloned in here?
The marker is 1kb DNA ladder from Thermofisher, there is a faint band at the bottom (~250bp). The control consist of the same DNA used for other lanes.
Update: I did the EcoRI digest, if only i could upload the gel here on replies but i cant. Well, i got my insert in lane 2,3 and 5 (although a bit faint). But i also got 3 weird bands on top of my insert which i cut with EcoRI (maybe my ligation did not go well, or some contaminants, or ratio of DNA vs Enzyme was not right). The control band which was not cut was ~4000bp.
Now that i have confirmed my insert, i was thinking of combining the remaining 3 samples which had my insert, streak a plate, take half of positive clones and go directly and do a double digest with NdeI and XhoI. The remaining positive clones on the plate i will take them for sequencing to confirm my insert.
Does this sound like the right way to go from here?
You should be able to attach an image, you can go through the "My Media" button, next to the emoji one in the reply box, or use the "more reply options" in the lower right corner, next to the post button.
Great that the cloning is working. The weird bands might be the result of star activity.
Typically it is not a great idea to combine individual colonies for this sort of thing as individual clones can have errors from the PCR step you used in the cloning process. I would take a few colonies, grow them individually in liquid medium, make a frozen stock and extract the plasmid for sequencing. Once you know you have a correct sequence, then I would grow up more of that particular clone and use it to do the rest of the process.
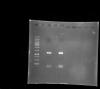
Managed to attach my gel. Lane 1 represent 1kb marker, lane 2 is control without EcoRI (2.5uL DNA + 6.5uL water +2uL Buffer). I then skipped lane 3 and loaded my samples from lane 4 to 9.
My insert are visible (though faint) at lane 5, 6 and 8 (~1197bp), if only u can enlarge the gel, u will see them. The rest of the bands on top are probably some sort of contamination or star activity as you put it.
Just for comparison, i have also attached by PCR amplicons gel (duplicate) confirming my DNA at ~ 1197bp.
Would really appreciate your response ones again.

Forgive me for asking too many questions. Now that i had identified that 3 of my samples contain my insert, how do i go about it streaking my plates again with this 3 samples containing pGEM-T and my insert? Am thinking too hard and i cant get around this.
It depends on what you have - if you have a plate with a small streak taken from a colony when you picked them for insert checking, then you would lift from that - you don't need to re-streak in this case, but you can if the plates are more than a week or so old, in which case the antibiotics may have deteriorated and re-selection would be a good idea.
If you have just the DNA - then you will need to re-transform, spread a plate and pick it.
If you have a frozen/glycerol stock, then you can just use that to streak out a fresh plate, or even directly inoculate a liquid culture.