PCR product not migrating from wells...?? - (May/14/2015 )
I'm working on a project involving a new protocol for skeletal DNA extraction (usually poor quality DNA in the first place..) and I am using agarose gels to visualize amplifications of a 100bp region we use in our lab to assess amplification success. Unfortunately, I'm getting some results that I've never seen with my gels before, and was wondering if I could get some interpretation help.
I'm using 1.7% agarose gels and 1x TBE buffer solution. Gels are run for about 25-30min, or until products have migrated to about half way down the gel. Ethidium bromide was used for staining.
For amps, each sample consists of 12.5ul EconoTaq, 6.5ul DNA-free water 1ul forward primer, 1ul reverse primer, and 4ul of sample. PCR is run for 40 cycles with an annealing temp of 48c.
1: Negative control (bone and DNA-free water without having going through the extraction process... water was removed and amped)
2-9: Test samples
10: PCR positive control (DNA extracted from blood, with known success)
11: ladder
12: PCR negative (DNA-free water instead of extraction sample)
I didn't expect anything to show up in wells 1 or 12, and the gels confirm my expectation. However, for wells 2-9, it looks like the ethidium bromide is staining something around the wells, along with a light smear continuing down each lane. What is going on here? Is it genomic DNA that isn't being amped by our primers? Our next step is to test another set of primers for a different STR and see if maybe that will amp something. Is our DNA too degraded? Is there a different angle we should maybe approach this?
Thanks for any comments/suggestions/insight. It's very much appreciated. If there is any other info you'd like me to add, just let me know.
Cheers,
Strand
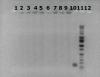
it appears that you did not have a significant amplification reaction. so, the bands on top are most likely your isolated dna.
in your recipe, i don't see reaction buffer listed (or is it part of the 12.5 ul taq?).
Standr,
it does not look like your PCR has worked for the samples. There is DNA there like mdfenko said, but is not being amplified by your primers.
Mdfenko,
The buffer is in the Taq, rather the Taq is in the 12..5 ul buffer that is being used. This is the Next Gen of scientists who hate pipetting individual components into a vial.
(Actually, I prefer something like for routine work). But when it comes to standardization, there is nothing better than the old school.
Ameya P on Fri May 15 13:00:09 2015 said:
The buffer is in the Taq, rather the Taq is in the 12..5 ul buffer that is being used. This is the Next Gen of scientists who hate pipetting individual components into a vial.
(Actually, I prefer something like for routine work). But when it comes to standardization, there is nothing better than the old school.
i thought that might be the case but when i looked it up the listing showed separate reaction buffers, not specifying that the taq was in them. thanks for the information (and the editorial comment).
I'd suggest that the gel indicates there is too much DNA in the reaction. This can be inihibiting. I doubt if you should be able to see genomic DNA from the template on a gel. You might try running the reaction with less (perhaps much less) template DNA. There could also be pcr inhibitors in your 4 ul of template added. Try diluting your template 100x with water.
I disagree about PCR master mixes and standards. PCR has too many variables as it is, and having non-uniform, inconsistent amounts of magnesium in your reaction, for example, is not empowering, but rather confusing. Master mixes just make sense for almost all applications, in my opinion.
Thanks for all the comments and suggestions.
That was our original suspicion, that the bands under the wells was DNA not being amped by our primers. Our next step is to try another set of primers targeting a different STR to see if that might work.
We don't think there is too much DNA in the reaction; we are dealing with low-quality, low-quantity DNA samples that are typical of most skeletal DNA extractions. We also don't think the DNA contains any PCR inhibitors. We have already established controls for each bone sample using a spin column extraction method that verified that DNA can be extracted from the bone samples and that our STR can be amped without inhibition. We are just testing a new extraction technique.
Side note: The buffer is in the EconoTaq. I'm one of these next gen grad students, and while I do enjoy pipetting (I honestly find it somewhat therapeutic to calculate amounts and add them carefully to tubes...), the master mix makes it easy for me to quickly set up consistent reactions. I'm thankful to have an advisor who values the old school methods; she teaches an undergrad/grad class where we do RFLP, VNTR, and some of the other analyses that have been used in forensic cases. And instead of simply plugging-and-playing and allowing comps to do all the work for us, we learn about each component of the master mix. Gels are not only efficient for us, but also allow us to appreciate the difficult role of interpretation. Fun stuff!!
in what buffer do you solubilize your template dna? if the buffer contains edta then you may be depleting the mg in the reaction with the large volume of template added (you might also be changing the pH enough to affect the reaction).