Please help uneven bands in western blot - (Jan/30/2014 )
Hi all,
I am new to western blot and currently I am working on detection of phosphoproteins. Please see the attached picture for the detection of total p38 proteins (which I use to normalize the phospho-p38 proteins).
May anyone suggest why there is uneven distribution in a single band and why there would be a difference in total p38 proteins in different treatments which theoretically should be the same?
And may I know if I can use GAPDH for normalisation of phosphoprotein instead of the corresponding total protein (since p38 cannot be probed after stripping so phospho-p38 and total p38 cannot be probed on the same membrane)?
I did the protein assay the same day as running gel. I quantified the proteins and boiled the proteins with sample buffer and then added 80 ug of proteins into each well. I had already tried to make sure that the sample buffer is well mixed without any visible precipitation, and vortex and pipette up and down before adding into the well. I ran the gel with 150V for 2 hours and transfer by semi-dry method with PVDF membranes. I had tried to eliminate the bubbles by rolling even layers with test tube.
Thank you very much for your help!
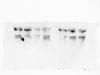
It seems to me that you probably have two problems, neither of which is bubbles in the transfer. However, I would say that your transfer is not working properly as you have some uneven smearing going on in some lanes, which could be due to the gel and membrane not being tight enough together during the transfer.
I would also say that you may have a problem with the removal of combs giving you an uneven well, which can result in what you are seeing. Make sure that your combs are clean before putting them in the gel.
It could also be that you didn't get rid of the precipitates that can form when you boil samples - your best bet here is to boil, cool on ice for a few minutes, then spin down for 30 sec or so in a bench top centrifuge.
For the transfer, I am using the ECL Semi-dry Blotters. May I know what can I do to ensure that the gel and membrane are tight enough together? I add weight above the blotter during transfer, should I add more or there is something else that I can do? I can also switch to wet blot if that would make better transfer.
Thank you so much bob1 for your reply and I will do it next time the same way as you recommended.
bob1 on Thu Jan 30 19:44:00 2014 said:
It seems to me that you probably have two problems, neither of which is bubbles in the transfer. However, I would say that your transfer is not working properly as you have some uneven smearing going on in some lanes, which could be due to the gel and membrane not being tight enough together during the transfer.
I would also say that you may have a problem with the removal of combs giving you an uneven well, which can result in what you are seeing. Make sure that your combs are clean before putting them in the gel.
It could also be that you didn't get rid of the precipitates that can form when you boil samples - your best bet here is to boil, cool on ice for a few minutes, then spin down for 30 sec or so in a bench top centrifuge.
To prevent the gel and membrane not being in close enough contact, you can add a bit more weight, and make sure that they are rolled together when setting up the transfer. It may also pay to ensure that you don't put too much liquid between the gel and membrane, but this isn't normally the problem.
I put more than 5kg on the transfer blotters (which some of my labmates even use more). But lately the buffer had ran out during the transfer twice so I add more buffer to the stacks, not sure if that created the problem.
Thank you very much for your advice and I will try to do it with the suggestions and see if it works!
I have tried out the suggestion and levels of GAPDH (loading control) and the total protein of i.e. p38/ERK still varied in the samples on the same membrane. I have used the coomassie blue to stain the total protein on a gel which was in the same run and there was no loading problem. May you suggest any possible reason for this?
Thank you very much!

Total protein stained with coomassie blue, each lane was loaded with 20ug total protein in 8ul

GAPDH 20ug total protein in 8ul

Total ERK 20 ug total protein in 8ul
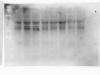
Total p38 60 ug total protein in 24ul
Coomassie is not very sensitive to amounts of protein - it will only detect 50-100ng/band, but blotting and probing with an antibody can detect much (much) smaller amounts and will show variation accordingly.
How did you quantify the proteins? Check the manual for reference range and curve fitting for your system.
Could you try to load based on cell number rather than amount of protein?
Antibodies also have the problem that if the solution isn't homogenous, or is unevenly incubated (i.e. one end not covered properly in solution) then you can get uneven staining, which may be part of your problem here.
So it means that the coomassie blue staining that I did actually cannot show I have loaded the same amount of protein in each lane?
I am using the Biorad DC Protein Assay with the standard curve using BSA of 0.125 mg/ml - 1.5 mg/ml. Usually the standard curve that I get has a R2 of 0.99. And I dilute my samples by adding 5 ul of sample to 45 ul of lysis buffer (1:10).
I think I cant load with cell number as I get only about 60 -70 ul of samples :(
My antibodies are prepared with 5% BSA or 5% milk in TBST, I incubate the membrane with the primary antibodies at 4 degree overnight with agitation and I reuse the antibodies. Do you mean maybe the antibodies are not evenly distributed?
Exactly - coomassie will not show slight differences in bands that might show up with other detection systems - hence the need to ensure equal loading with a normalization protein.
If your samples lie outside (above or below) the reference range (the manual says to use 0.2-1.5 mg/ml) then extrapolation is very difficult, as how the curve (and it is a curve, not a straight line!) behaves is not shown. Biorad is terrible for this sort of thing - they should tell you what sort of equation to use for the protein estimation. Also ensure that your lysis buffer is appropriate for using with this kit! Mercaptoethanol prevents the kit from working properly as do a number of detergents.
To base on cell number, you need to lift, count, then lyse at a certain density. I use 10^4 cells/ul and load 10 ul, and that is usually enough for most proteins.
Uneven incubation happens when there isn't enough liquid surrounding the membrane, so parts of the membrane don't get exposed to the antibody enough. For a typical membrane size a minimum of 5 ml is required in a tray or bag.
also, reusing antibody solutions depletes them (each use has a lower concentration). at some point they will be insufficient.
we stopped reusing antibody solutions so that we would obtain consistent results (doing this depends on the amount of antibody you have and use).