PCR product running on agarose gel - (Feb/20/2013 )
Dear all,
I would like to ask about a problem that has come to me when analyzing the agarose gels I have performed with my PCR products.
I´m trying to find the optimum annealing temperature for my pair of primers, therefore I have conducted a PCR reaction for each of my pair of primers and have visualized them in an agarose gel.
The problem is that the lanes I obtain in the gel, even if they are strong and clear, are smaller than the lanes obtained from the DNA Molecular Weight Marker that I use (DNA Molecular Weight Marker X 0.07-12.2kbp). Therefore, I´m not sure if the lanes I´m looking for in the gel are the result of primer dimers or in contrast, they are the result of a specific PCR reaction, where the obtained DNA size is small.
This image is an example of the lanes obtained in one of the runned gels.
Whereas, in the next figure (other pair of primers that analyze the same sample) two aspecific lines of lanes could be observed.
I would really appreciate if someone could help and give me any advice in relation to this, as these PCR products that I want to obtain will be to send for next generation sequencing once purified.
Thank you very much in advance for your help,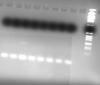
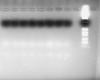
To me the lowermost band in both the gels looks like primer dimers. btw wats the product size?? if i am right your markers lowest band is around 70 bp, if so the bands that are seen are far below to conclude them as dimers, anyways you got to know the size of ur product to confirm it.
The first pair of primers are dimers. Go to https://www.idtdna.com/analyzer/applications/oligoanalyzer/ and type the sequences of your primers in there and you will likely see a high probability of dimer formation.
The second gel has very faint bands around 100 bp? That will be tough to sequence alone since it is so short. You need to figure out what the expected product size is and redesign primers. Also, if you are looking for a 100bp product, you should not be using a 12kb ladder and should increase the agarose % to create better resolution.
Thank you very much both.
The problem is that I don´t know which should be the size of my PCR product, how can I figure out? On the other hand, I won´t be able to redisign my primers as these ones are the ones suggested by the enterprise that will perform the sequencing analysis, so, do you think that I can start changing other conditions such as primer concentrations for example?
Thanks
unless you know the size and source of your product your proceedings wont help you much in standardizing your PCR. If you know atleast the sequence of the primers (usually written on the primer vials ) you can do a primer blast on NCBI (unless they are not the commercial primers), even here you shd know the source of the template you are using inorder to interpret the possible product. i think you have done a gradient PCR with two different primers against the same product?? wat gradient you have used? in the first gel and 3rd lane there looks something like pcr product, if tat is your actual product stick on to that annealing or around that and try altering the primer concentration, but looking at the dimer (??) levels it looks unlikely to avoid there, if atleast you could enhance the product and decrease the dimers you can do a gel extraction and sequence..even in the second gel all the lanes showing a band (hazy though) there, but the band intensity doesnt impress me..
Thanks GNANA. You´re right I have done a gradient PCR with two different pair of primer against the same product. I have proved different annealing temperatures, in that case, the máximum temp was 60 degrees estarting from the left hand the minimum 50 degrees. On the other hand, from by primer sequences I have seen that the product length that I should expect is ~ 560 bp, but I´m worried about being able to obtain such a big size. I have used 1 microM primer concentration (the maximum suggested by the pfu polymerase protocol), may be I could try reducing it to 0.2 microM concentration and increasing a little bit the DNA concentration....once again performing a gradient PCR..what do you think about that?
Thanks a lot,
I think you arrived this gradient based on the tm of your primers?? then PCR'ing 560bp is nowhere near to be worried. yes decreasing primer conc may decrease your dimers, and that wnt help if the primers are more potent to anneal themselves than to the template. 0.2 micromolar looks reasonable is your case. you can also reduce Mg concentration. Another tip, keep the reaction mixture on ice until you place it in the cylcer alternatively HOT START also would help. check out if your pol is compatible to do hot start pcr, as some may come with inactivating tags that needs the initial denaturation. 100ng (if ur template is genomic DNA) shd be optimal for PCR.
I have made another try with 0.2 mIcroM primer contration and 3 ng/microlitro of DNA concentration (the maximun recommended by the pfu polymerase protocol that I´m using is 10 ng/microlitro). I have made a temperature gradient once more from 60 degrees to 50 degrees. However, this is what I have obtained. I know the image is not clear and the ladder I´m using doesn´t help too much, but don´t think the lane showing the "bands" are near 500 bp (which is the expected size), what do you think? I´m not sure how to read the size of that lane...I have attached the reference of the DNA molecular weight marker I have used (I will try to use a better one next time),just in case you could give any suggestio.As it could be seen primer dimers that were observed in the first images are not so visible now, but I don´t know how to do to improve the PCR and obtain a clear band....
Thank you very much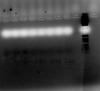

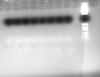
what is your template? is it genomic DNA? how much DNA you used? you have said 3ng/microlitre DNA but that doent answer how much you used / reaction?
I use 2 microlitres (6 ngs) in 50 microlitres of reaction, and it is DNA extracted from faeces samples, genomic DNA.