ChIP input - sonication problem? - (Aug/05/2012 )
I was doing CHIP for the first time with a lysate that was sonicated by my colleague. She did CHIP before and got some results (first with some custom protocol, second time with Imprint kit from Sigma).
As arranged I did additional MNase treatment on the 4 lysates (1U of MNase for 500 ul of lysate, as mentioned in Sigma manual, except for 5 instead of 2 minute incubation). This was the same protocol as she used before.
I then did the immunoprecipitation following the Imprint kit manual, de-crosslinking and DNA isolation. Problem was two of the inputs were negative in the initial real-time test, the other two with Ct around 37. Most CHIPed samples were negative too, but some also somewhere around 35-37, even some of the samples, that were negative as inputs.
I repeated the assay again, with same lysate but used Qiagen columns instead, with bit better, but qualitatively the same result.
I put the lysates on 1% gel to see how much it got fragmented before and after MNase treatment. I put there 5 ul (first picture) and 10 ul (second) of each lysate, either crosslinked to compare or quickly de-crosslinked by following protocol: dilute 1:1 in TE buffer, 10 minutes 98°C.
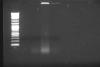
(lines: 2-log marker, samples 1-4 MNase treated de-crosslinked)
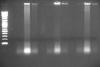
(lines: 2-log marker, samples 1-4 sonicated-only crosslinked, samples 1-4 sonicated-only de-crosslinked, samples 1-4 MNase treated crosslinked)
Only samples that had positive inputs were 3 and 4.
The concentration of lysated cells was supposed to be 1.6 for samples #1 and #2, 3.4 for sample #3 and 0.7 for sample #4 (all of it x10^6/100 ul).
So there is evidently not the same amount in 1 and 2 even if it should be, and actually seems to be more of sample 4 than it should be. The crosslinking seems to have efect mostly on the brightness of the background.
I have two questions:
1. Is the quick decrosslinking method mentioned here sufficient for just checking the length of fragments or it can skew the image?
2. In my unexperienced opinion, this looks like far more fragmented than it should be, with fragments mostly pretty shorter than 100 bp. But MNase treatment didn't seem to mess this up, as it looks fragmented same as in the sonicated-only samples. Or maybe degradation of the lysate prior to sonication, it spent a year on -80.
So seems I have to do a new sonication, right? This time maybe in shorter steps, and check it along the way (for this I would like to know, if I can de-crosslink it with just boiling for 10 minutes)
The number of cells put into each immunoprecipitaton was adjusted to be the same for each sample, approx 0.42x10^6 per well comming close to the upper limit recommeded for one well, the input was 1:20 of this amount.
The reason for samples 3 and 4 being the only positive inputs it's probably in the nature of samples, they are mouse-human hybridoma cells, with variable pecentage of human chromosome, primers only detect human DNA, samples 1 and 2 have lower percentage than the control samples 3 and 4. Anyway the main problem is I can't put more lysate into CHIP since that would exceed the recomended limit, but I think I have such low (or none) Cts, because my DNA is too much fragmented from the start, that I'm detecting only the scarce longer fragments (real-time amplicon length is around 100 bp). Right?
It appears to me that your DNA is over-sonicated. On the gel, I only saw very low molecular weight DNA (well below 100 bp). Here are my suggestions:
1. why don't start with fresh cells instead of lysate prepared by others and stored at -80C for one year? Of course if you don't have the cells anymore, there is no choice. For a new user of ChIP, it is better to start from scratch.
2. Before you do actual IP, you need to optimize your sonication conditions by reverse crosslinking your sonicated samples and running a gel.
3. Is your PCR primers working fine? You need to use genome DNA as positive control to see if the primers work at all and determine their amplification efficiency by real-time PCR.
1. That is pretty difficult to do for many reasons, however I understand it can be a problem.
2. Yes I know, is the quick reverse crosslinking protocol mentioned above sufficient for such gel check?
3. Primers are fine, all this was done.
My PI reviewed results today and said it's a degraded lysate, not oversonicated. The only way is to repeat the cell experiment again.
I agree with your PI, there is a concern of DNA degradation. Q
This kind of test is wrong.
You DNA did not move in Gel at all.
First centrifuge you sonicated sample, then keep the supernatant which is your sample from now on.
You should take 25 microlitter of sample.
Then add 2 microlitter of Proteinase K + 2 microlitter of RNase cocktail
First 30min in 37 degree centigrade (vortex and spin each 10 min)
Then 30 min and more in 65 degree (vortex and spin each 10 min)
then load it on Gel 1% and less.
Bound-Proteins do not allow to your DNA to move in Gel.
I have checked some quick decrosslinking, but now I can say that this way that I say is the best for checking the DNA.
-----
Babak
My $0.02 worth is that what your actually looking at on your gel is RNA and your lysates shouldn't be degraded after a year or so in the -80. I have ChIP'd lysates (granted for histone marks) well over a year after the lysate was frozen........for what its worth your sonication looks pretty good (picture 2 middle lanes).........your reverse crosslinking in TE is fine.......maybe try longer, but definitley add at least RNase for a bit before yoou load it onto the gel.