Odd gel run following PCR - (Jun/28/2012 )
Sure.
Aenaturation
Temp: 95
Time: 20 sec
Annealing
Temp:63-71
Time: 1 min
Extension
Temp: 72
Time:1
In addition my stock concentrations are below:
DMSO (100%)
10x Bufer (10x)
MgCl2 (25 mM)
dNTP + deanzaGTP (2mM each)
Forward (10 uM)
Reverse (10 uM)
TAQ (5 units/uL)
Hi Veteran, thanks for the tip about cycle number adjusting. However if my band is faint shouldn't I increase my cycles from 40 to about 45ish?
40 cycles are way too many already. more cycles only mess with your results further. I would use more template per reaction.
princeofnam on Mon Jul 2 16:31:13 2012 said:
Hi Veteran, thanks for the tip about cycle number adjusting. However if my band is faint shouldn't I increase my cycles from 40 to about 45ish?
If a PCR isn't getting you a good product by about 30 or so cycles- then there is something wrong with some aspect of your reaction. You are better to optimise the PCR rather than simply increase the cycles, which is a quick "fix" that may not work, and doesn't address the actual problem in your reaction.
Putting your reagents through excessive cycles will diminish them, and your polymerase will start to lose activity. Also I've been told that the likelihood for errors increases.
You said you checked your DNA template by spec- so can you tell us how much DNA you add to each reaction- that should tell us whether the amount of template is the problem (highly unlikely, it is almost never the amount of template that is the issue).
I know you said you didn't want to run a dilution series with one of your templates (or you supervisor said not to) but don't forget too much template is a PCR inhibitor.
Here is what I would do:
using the template that got you the faint band in your second attempt:
1. Do a dilution series, and set up PCR tubes for neat, 1/10, 1/100 and 1/1000
2. Add master mix (using only 1ul of primer rather than 2.5ul)
3. Run the PCR with your 40 cycles (but I would probably reduce the annealing time to 15-30 sec), and I would also reduce the denaturation temperature to 94C (this is optimal for taq). You could also check the product insert for your taq to see what they recommend.
4. Use the annealing temperature that worked for your successful reaction above
Thanks Veteran. Before I could run a dilution series my mentor wanted me to run my PCR on a primer gradient first from .5-1.5 ul/well.
Btw, usually I add 49 ul of mastermix w/ 1 ul of DNA to each well. The primer gradient I set up previously turned out as follows. 
Well 1: 5 ul 1 KB plus ladder
Wells 2-3: .5 ul primer
Well 4: NC w/ .5
Wells 5-6: .75 ul primer
Well 7: NC w/ .75
Wells 8-9: 1.25 ul primer
Well 10: NC w/ 1.25
Wells 11-12: 1.5 ul primer
Well 12: NC w/ 1.5
My mentor suggested I run it at more cycles contrary to your advice actually and we ran it for 42 cycles.
In summary, 42 cycles, 95 C, 30 sec, 55, 30 sec, 63.1 30 sec, 72 C, 1 min
I actually don't know why the 55 30 second period precedes the ideal temperature I calculated and why I shouldn't just run the PCR annealing temp at 63.1 for 1 min. My mentor has always set the PCR reaction up with the 55 30 sec annealing period so I left it that way. I will have to ask her about that the next time I speak with her. One problem I did notice are the presence of multiple bands (which shouldn't happen in my PCR reaction) and some smearing. Could this be because the number of cycles as you mentioned is too high? or is this more indicative of having to do a dilution series you think?
I think that the multiple, non specific bands are due to you using a 55 annealing temperature as part of your cycle. I really don't know why your mentor is suggesting that as part of the cycle. It makes no sense at all to use a lower annealing temperature than required for a specific product as all you do is encourage non-specific binding and amplification.
Additionally, unless I am reading this wrong, your cycle goes:
denaturation, annealing 55 , annealing 63.1, extension
what would happen here is that in the first 30 sec at 55 all of your annealing would be done anyway- so the 63.1 would be pointless...no wonder you are getting non-specific binding.
Ask your mentor for the reason you are using the 55C.
Also, 1 minute for annealing time is WAY too long. Most cycles use 15 to 20 seconds, or even 30 seconds max. But 1 min is overkill.
It is encouraging to see that the amount of primer dimer has drastically reduced, so now you know you can use far less than you were originally.
I know that you obviously have to follow the advice of your mentor, and that you are at her mercy. But to recommend adding more cycles when you are already at 40 is, in my opinion, really bad advice. Yes in theory adding more cycles should allow a faint band to become clearer- but it doesn't address the fact that the PCR itself is obviously very bad if you need this many cycles.
And I am at a loss as to why she won't allow you to do a dilution series with your template. The smearing that you are seeing suggests to me that you may have too much template in your reactions. What is your template, by the way?
(by the way, when you showed your mentor your original photo, what did she say about the bands at the bottom?)
Let me know how you go with this, we'll sort it out 
What LeeLee said.
Sorry to come back with a reply so late. I had already scheduled to go home for the summer, sadly right in the middle of my trying to figure out my PCR. I attempted another PCR with the same settings as above but with a few modifications
1) I dropped down to 40 cycles from 44
2) I ran a PCR Temp Gradient from 58-63 C
3) I took out the 55 C step.
4) Each well had 1.25 ul of forward and reverse primer each.
I'm wondering if the 55 C step was originally in my saved protocol because I started with a temp gradient protocol. Do you require an initial lower temp for temperature gradient runs? I'm also unsure about the giant black spot on my PCR. I any event, lanes 3-11 are my temperature gradient from 58-63. Lane 5 is my control. Lanes 3, 7, 9, 11 are control 1, and Lanes 4, 6, 8, and 10 are another control2 . There seems to be a consistent band for Control 1, although you can sometimes see it in Control 2. As always the bottom bands are indicative of rampant primer dimerization which I haven't figured out yet. There at least seems to be bands from non-specific binding as indicative from my previous post, although I'm not sure either if this one band I'm seeing is non-specific too. One of the bands I'm looking for is ~611, although other VNTR polymorphisms might be present in 48 bp intervals. Pretty lost as to what my next run should be. Perhaps higher annealing temperatures? Lower primer amounts? Although there didn't seem to be that much of a difference with primer amounts below 1.25 ul in my primer gradient run.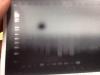
If I am understanding you correctly, you are doing a touchdown- not a temperature gradient.
A gradient means that each reaction tube gets only one annealing temperature...so say you have 8 places that you could put your PCR tube into- the first one would be at 58C, the last at 63C and the machine would do a gradient of temps between those two for the rest.
Eg.
58....59.....60....60.5.....61......61.5......62......62.5......63 (or whatever, it would tell you what the temps are).
What you are doing is a touchdown- where during the run each tube has a few cycles at each temp as the machine runs the cycle from your first temp (58) to last (63). I usually actually run touchdowns the other way (from high to low) but I don't know that that would matter.
In any case, you have removed a great deal of your non-specific binding as the only band present is your desired band (plus primer dimer of course). The smearing suggest to me that you are still running too much template.
Have you quantified your template? How much are you adding to each reaction?