Restriction Digest Problem - (Jan/26/2012 )
I have been trying to digest my pET32a vector with both EcoRI-HF and XbaI. The restriction sites are approximately 550 bp apart from one another. I have tried 2 hr, 4hr, and overnight digestions. Below is my reaction mixture.
pET32a vector (3 or 1 µg) 28 µL
NEB Buffer 4 5 µL
BSA (100 µg/ml) 0.5 µL
EcoRI-HF (40 units) 2.0 µL
XbaI (40 units) 2.0 µL
H2O 12.5 µL
TOTAL 50 µL
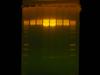
Attached is the gel image. Lanes 2 and 3 are EcoRI only for a linear control. Lanes 4 and 5 are 3 ug vector double digest, and lanes 6 and 7 are 1 ug vector double digest. All digests were overnight. You can see that the correct size fragment is present around 600 bp (200 to 12000 bp ladder) but it is very incomplete
Does anybody have an idea as to why I have incomplete digestion? I think it might be the plasmid prep. The uncut plasmid hardly migrates on a gel as shown in the Picture 1 figure in lane 2. The plasmid could be nicked, and that is why it wont migrate but it still should be digested.
It seems as if no matter how much enzyme I add or how long I digest, I do not get any significant digestion.
Any advise? Tomorrow I will be doing a new plasmid prep.
Thanks
It could be the prep - how did you do it? Carry over of bacterial components in plasmid preps are the most common cause of restriction enzyme inhibition.
I used PureYield Plasmid Maxiprep System from a glycerol stock. Everything seemed to go smoothly when I did it, no roadblocks, but it looks terrible on a gel and I may have wasted a couple weeks trying to use this for cloning.
More than half of your reaction volume is DNA. This is a recipe for DNA inhibitors in your prep to cause problems. Try again with a 5x dilution of your DNA (That is, do a digest in a volume of 250 ul rather than 50 ul).
Inhbitors can come from many places, but likely ones are ethanol from the washing step and guanididium from the binding / lysis step. But you don't care -- dilution is your friend.
Thanks phage. If the digest is successful, which it should be in an hour ideally, should I see no traces of uncut plasmid? Or is that too ideal and I just need to make sure I have good separation on my gel so I can extract the proper band?
You could also have very high genomic DNA contamination in your prep, which would show as DNA but not be cut with your enzymes. Make sure you are not vortexing or carelessly handling the DNA after lysis and before clearing the lysate.