BN-PAGE of purified membrane associated protein - (Nov/16/2011 )
Hello everyone,
Since native page never worked with my protein I started to run BN-gels as it is supposed to be a good tool for hydrophobic proteins. I'm using gradient gels (3-16 %) because the protein seems to form high aggregated complexes.
There is still one problem. I got protein smears so that I cannont perform Western blots. Attached you find an example. In the first two lines I loaded two different markers; first is BSA, second JackBean Urease. Then different protein samples follow. As you can see only BSA makers shows a nice distinct band.
I found several protocols and tried so far the one from Wittig et al (https://www.nature.com/nprot/journal/v1/n1/full/nprot.2006.62.html) and lately the one attached.
Since I have no real experience with native pages I would appreciate any suggestion to get rid of the smears.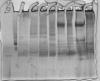

have you tried to blot the gel?
there appear to be reasonably well defined bands in the "smears".
smears often indicate incomplete solubility of the protein. you may want to clear the samples (by centrifugation) immediately prior to loading the gel.
are you using native gels because you want to maintain enzymatic activity? conformation? why not sds-page (bn-page is good for size separation)?
which other native page protocols have you tried? neutral pH, acid pH, basic pH (ornstein-davis)?
Yes, I tried to blot the gel and the complete smear is stained, and I used the half of the antibody than usual.
I'm doing native gels for conformational studies. So far gel filtration was unuseful, the protein was always found in the void volume. Then, I did DLS measurements and it was shown that the protein forms huge aggregates but is still stable. I started the BN-PAGE because the protein is membrane associated and very hydrophobic and BN-PAGE is supposed to be a good tool for such proteins.
Other Phd student used to perform native PAGE but the protein sample never entered the separating gel (10 % Ac). Now I found out that's due to the aggregates. Last week I started to repeat common native gels using a gradient from 4-16 %. The pH of the separating gel is 8.8 and the one of the stacking gel 6.8. The running buffer is 8.3. Now I see less smears although I don't like the resolution. I think the advantage of BN-PAGE is that the samples arer charged allowing size separation and ignoring the intrinsic charge of native PAGE.
So, there is still enough to play around. In general I would like to determine the oligomeric state of the protein vs. some specific mutants. Apparently it's not that easy 
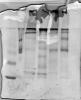
well, you said it yourself. the problem is that your protein aggregates. bn-page won't break up the aggregates.
is your protein aggregated immediately upon isolation or does it occur over time?
if it takes time to aggregate then you must run your gels as soon after isolation as possible.
we found that inclusion of 0.5-5% peg-8000 (also known as 6000) helps retard the aggregation of (at least some) membrane bound proteins. however, peg will disrupt the banding pattern in page, especially in the lower molecular weight range.
That's right. I did BN-PAGE to check several purified protein samples if they show the same aggregates, and I wanted to know the size of them. I've read a paper where they used BN-PAGE "get rid" of those aggregates. First they checked their protein then they started using several detergents and chekced again until they found one where they got a single band. And then they knew which puffer they need to get the crystal structure of their protein. Basically that's what I intend to do as well (addionally to 4D structure).
I guess it aggregates right after purification because last time I purified the protein, added sample buffer and ran the gel the next day (samples were frozen at -20 °C).
Since the protein samples ran with much less smear in Native PAGE I might go back to that, depending on the experiment. Last week I did some membrane preparation to isolate the membrane bound form of my protein and therefore BN-PAGE was much better.
So, there are many things to experiment with, yet. I hoped I could get rid of at least some "smear" by changing the protocol a bit here and there but that hasn't worked so far. But I will see want happens when I start to experiment with some detergents...
Where do you add PEG-8000? To the sample?
I really appreciate any idea...it's better to try things out than have nothing to do
So thanks unitl now.
we add peg to the sample (it's a component of the final purification buffers).
some proteins don't like to be frozen. you may want to try running the page immediately after isolation.
from what you wrote, the bn-page was not the disaggregating factor, the detergents were. the bn-page was used to determine which detergent was effective (and could have been performed using native page).
yes, experiment with detergents to find one that will maintain the disaggregated form of your protein. we found peg, although not a detergent, was best for our protein. you should find what works best for yours.