Problems eluting a GST fusion protein in GSTrap FF - (Nov/09/2011 )
Hi guys,
I am trying to purificate a 50 kDa protein, which has no cysteines, fused to GST. This construct has been previously expressed with the GST protein in other labs, even a lab has managed to cristallized the protein once scised by thrombine.
All the cloning steps were succesfull in my case. Nevertheless, I am expericing problems in the affinity chromatography step...After Lysozime-EDTA or sonication treatment, the fusion protein is mainly in the supernadant (even after filtration through an 22 um filters). After loading it into the GSTrap FF 5ml there is almost no fusion protein in the flow through. The point is that in the elution step there is very little amount of fusion protein (not more that a couple of mg).
I have previous experience in the GST fusion system as I had been succesfull in the purification and characterization of an human protein before... This time, I have tried to maximize the elution step increasing even to 150 mM reduced gluthatione, 500 mM NaCl, pH 9,0 but nothing changed..
Looking at the photo could it be that the fused protein still bound to the GSTrap? Could it be ta good idea to perform a batch purification with sepharose 4B instead of using GSTrap FF?
A: supernadant after cell lysis (diluted ¼ in the gel)
B: sepernadant after cell lysis and filtration (diluted ¼ in the gel)
C: flow through (diluted ¼ in the gel)
D: elution of fusion protein (diluted ¼ in the gel)
I would be thankfull if you could give some advices because I am becoming crazy..
Kind regards,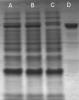
I don't get it. Why do you expect more protein? What about the other elution fractions (or do you elute in one fraction; ml?). How long did you express your protein in how much medium?
I expected much more protein because the induction takes 3-4 hours and from a 1 L LB culture, I recover around 2 mg (and the fusion protein is 75 kDa). When I used to do the same with another fusion protein (26 kDa GST+ 4 kDa my protein= 30 kDa) I used to recover more than 10 times fusion protein in mass (30 vs 75!)! and the induction is as good as it was with that protein... 
It's not unusual that one protein is purified in a much higher yield than another. How do you know that the induction is similar (in total amount)? Have you tried to increase the yield (e.g. 2 L culture or use another medium like terrific broth or induce the expression at higher OD (e.g. 0.8 or 0.9). Then I would expect that you get more protein. Otherwise there is really something with your column. Maybe a part of your protein precipitated at the column. Are you sure that the buffer is good for your protein?
Somebody told me about that medium TB and I never heared before...Is that a preferred medium for recombinant protein expression in E. coli?
I do not now if the buffer is ok for my protein and the induction is similar according the SDS-PAGE gels!!
It's preferred in that way that it's a rich medium which allows higher production. I prefer to use it especially when I express at low temperature.
If the bands are similar then you have to keep in mind that when a small protein shows a similiar band (in terms of intensity) than a big protein there is much more total amount of small protein than the big one. Is it clear what I mean?
You wrote that your protein was always found in the supernatant, right? so you can exclude that your protein in not as soluble as you other one.
If I were you I would try to increase induction time first...lets say over night at lower temperature. Then check the induction, the supernatant and the pellet and perform the purification.
You can also check if you can add some special buffer to remove everything stuck to the column material and look if you can find your protein there. Unfortunately I cannot remember what I used to clean my gel filtration column. I think it was a NaOH solution but don't know the concentration. At that time I had the issue that my protein precipitated onto the column.
Good luck!!
thank you for the advices!