Smearing on gel in PCR products - (Mar/07/2010 )
Hii
I am trying to amplify a microRNA sequence from Brassica genomic DNA. Initially I got a single sharp band with Biotools Taq Polymerase and also through Fermentas Pfu polymerase. However, when I repeated the experiment for purification of the product, I wud get a lot of smearing on the gel. I repeated the PCR with a number of modifications. I checked the DNA stocks for degradation also but the they were fine.
I tried reducing the amount of template and dNTPs, however the smearing problem is not being overcome.
What can be the possible reasons?
Anuradha on Mar 7 2010, 11:42 AM said:
I am trying to amplify a microRNA sequence from Brassica genomic DNA. Initially I got a single sharp band with Biotools Taq Polymerase and also through Fermentas Pfu polymerase. However, when I repeated the experiment for purification of the product, I wud get a lot of smearing on the gel. I repeated the PCR with a number of modifications. I checked the DNA stocks for degradation also but the they were fine.
I tried reducing the amount of template and dNTPs, however the smearing problem is not being overcome.
What can be the possible reasons?
If your initial PCR rx produced a nice sharp band, you've got good primers, the right template concentration, and annealing temps for your reaction. There are many reasons for smears and different types of smears mean different things. When troubleshooting it is best to change one variable at a time instead of several at once.
A couple of questions:
1) Are you using the same thermocycler as you did for the successful run?
2) Are you getting fuzzy bands or long smears - a picture would be great
3) How are you storing your gDNA? 4C or freezer?
A good place to start is with your template DNA. Make a fresh prep and repeat your original reactions with original conditions.
Be sure to thaw your reagents completely and vortex before using especially MgCl.
Use the same thermocycler that you used the first time, use fresh running buffer, and make fresh agarose (agarose kept in water baths can get funky and run wierd)
You could also set up a second reaction with the fresh gDNA but use fresh reagents (water too), dNTP's, and polymerase.
These are just some basic things to start with, and there are many more things that could be going on.
Denny on Mar 8 2010, 04:39 AM said:
A couple of questions:
1) Are you using the same thermocycler as you did for the successful run?
2) Are you getting fuzzy bands or long smears - a picture would be great
3) How are you storing your gDNA? 4C or freezer?
A good place to start is with your template DNA. Make a fresh prep and repeat your original reactions with original conditions.
Be sure to thaw your reagents completely and vortex before using especially MgCl.
Use the same thermocycler that you used the first time, use fresh running buffer, and make fresh agarose (agarose kept in water baths can get funky and run wierd)
You could also set up a second reaction with the fresh gDNA but use fresh reagents (water too), dNTP's, and polymerase.
These are just some basic things to start with, and there are many more things that could be going on.
1) I am using the same thermocycler for all my PCR reactions.
2) I am getting a long smear along with some fuzzy bands sometimes (even when keeping all the reaction conditions as constant)
3) I am storing the gDNA and the working stocks at -20 C.
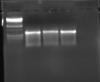
Also I am attaching two pics. One is my initial reaction where I was getting a single sharp reaction. Other one shows the smearing.
I tried to change the parameters one by one, however I didnt get any satisfactory results as yet.


Anuradha on Mar 8 2010, 09:11 PM said:
Denny on Mar 8 2010, 04:39 AM said:
A couple of questions:
1) Are you using the same thermocycler as you did for the successful run?
2) Are you getting fuzzy bands or long smears - a picture would be great
3) How are you storing your gDNA? 4C or freezer?
A good place to start is with your template DNA. Make a fresh prep and repeat your original reactions with original conditions.
Be sure to thaw your reagents completely and vortex before using especially MgCl.
Use the same thermocycler that you used the first time, use fresh running buffer, and make fresh agarose (agarose kept in water baths can get funky and run wierd)
You could also set up a second reaction with the fresh gDNA but use fresh reagents (water too), dNTP's, and polymerase.
These are just some basic things to start with, and there are many more things that could be going on.
1) I am using the same thermocycler for all my PCR reactions.
2) I am getting a long smear along with some fuzzy bands sometimes (even when keeping all the reaction conditions as constant)
3) I am storing the gDNA and the working stocks at -20 C.
Also I am attaching two pics. One is my initial reaction where I was getting a single sharp reaction. Other one shows the smearing.
I tried to change the parameters one by one, however I didnt get any satisfactory results as yet.
Hi Anu,
have u tried raising the annealing temp??? and also, I think its better to store the genomic DNA at 4 degrees than -20 degrees to avoid shearing caused by regular freeze-thaw cycles. And as Denny suggested, use a fresh aliquot of gDNA and other reagents (MQ, dNTP etc).
DRN on Mar 8 2010, 11:09 AM said:
Anuradha on Mar 8 2010, 09:11 PM said:
Denny on Mar 8 2010, 04:39 AM said:
A couple of questions:
1) Are you using the same thermocycler as you did for the successful run?
2) Are you getting fuzzy bands or long smears - a picture would be great
3) How are you storing your gDNA? 4C or freezer?
A good place to start is with your template DNA. Make a fresh prep and repeat your original reactions with original conditions.
Be sure to thaw your reagents completely and vortex before using especially MgCl.
Use the same thermocycler that you used the first time, use fresh running buffer, and make fresh agarose (agarose kept in water baths can get funky and run wierd)
You could also set up a second reaction with the fresh gDNA but use fresh reagents (water too), dNTP's, and polymerase.
These are just some basic things to start with, and there are many more things that could be going on.
1) I am using the same thermocycler for all my PCR reactions.
2) I am getting a long smear along with some fuzzy bands sometimes (even when keeping all the reaction conditions as constant)
3) I am storing the gDNA and the working stocks at -20 C.
Also I am attaching two pics. One is my initial reaction where I was getting a single sharp reaction. Other one shows the smearing.
I tried to change the parameters one by one, however I didnt get any satisfactory results as yet.
Hi Anu,
have u tried raising the annealing temp??? and also, I think its better to store the genomic DNA at 4 degrees than -20 degrees to avoid shearing caused by regular freeze-thaw cycles. And as Denny suggested, use a fresh aliquot of gDNA and other reagents (MQ, dNTP etc).
Hi Anu,
The gel pictures are very helpful. From what I can see, your original gel has faint bands that may have been better visualized on the second gel. Look at your ladder on the first gel, it's pretty faint too!
I agree with DRN and would try optimizing your annealing temperature using your primer Tm's or gradient PCR. Also check your extension time, is it enough for the polymerase your using and # of base pairs.
To add to DRN's suggestion on gDNA storage. When you make a prep and plan on using it in the next few weeks, aliquot it out into several tubes, store your working stock in the fridge, and your extra in the freezer. Once you thaw one, leave it thawed.
Sometimes certain gDNA preps just don't like hanging around the fridge for very long so don't panic if your pcr stops working, just grab a fresh tube out of the freezer.
I've also worked with lab's that swear by the same practice with your dNTP's. They claim that the freeze thaw eats away at the ends and makes them less efficient. I've adapted that practice and don't have any problems. Good Luck

P.s. Run your gels longer for better separation, your product has barely moved down the gel and your ladder is all scrunched up at the top. 