RE digest problem - (Oct/01/2009 )
Hi.
I had cloned an insert of interest into a plasmid vector which has the size around 2.9kb. The insert has around 4kb. I had picked up the colony and do a plasmid extraction from it. The thing is, I want to confirm that I had the correct insert into the plasmid vector. I am doing a sequential restriction enzyme digestion to check on it. The RE enzyme that I am using are (AscI, NotI) and (EcorI, HindIII). I did for both set for sequential re digest, but compare to an undigested plasmid vector with the insert; the gel image of the product of after digestion and before digestion shown at the same size-which is around ~7kb.
I am not sure if it is because I put too less of enzyme to make the RE digest. I only pipette in 10U and the digestion time around 16 hours.
What should I do to have it successfully cut?
Please advice and I am gratefully appreciate any suggestion and opinion.
Thanks in advance.
Cheerioet
Hmmm... a picture of your gel would be useful
Could you write out your the restriction digest formulation that you used. How much of each component did you add, and at what temperature. How much DNA are you cutting? (Might you have accidentally used TE to make up the digestion cocktail)
NOTE:: Your restriction enzyme comes a glycerol based buffer. Glycerol in is inhibitor of enzymatic activity. The total volume of enzyme should not exceed 5% of the digest volume, otherwise you would start experiencing enzyme inhibition and star activity. In other words more is not better for a restriction digest.
Could you also tell us what size fragments that you expect from each restriction digest? And where it cuts in your vector-insert molecule.
You might also what to clean up your DNA again.
Should a DNA sample be dirty, (ie containing one of many enzyme inhibitory compounds) the DNA won't cut. In such an event, the DNA sample needs to be cleaned. A phenol/chloroform extraction would do the trick.
perneseblue on Oct 1 2009, 06:35 PM said:
Could you write out your the restriction digest formulation that you used. How much of each component did you add, and at what temperature. How much DNA are you cutting? (Might you have accidentally used TE to make up the digestion cocktail)
NOTE:: Your restriction enzyme comes a glycerol based buffer. Glycerol in is inhibitor of enzymatic activity. The total volume of enzyme should not exceed 5% of the digest volume, otherwise you would start experiencing enzyme inhibition and star activity. In other words more is not better for a restriction digest.
Could you also tell us what size fragments that you expect from each restriction digest? And where it cuts in your vector-insert molecule.
You might also what to clean up your DNA again.
Should a DNA sample be dirty, (ie containing one of many enzyme inhibitory compounds) the DNA won't cut. In such an event, the DNA sample needs to be cleaned. A phenol/chloroform extraction would do the trick.
Dear Perneseblue,
Thanks for the fast reply. I am going to try a phenol/chloroform clean up before continuing the 2nd digestion.
I am using the followings protocol and formula.
dH2O 10uL
10X buffer3 2uL
10X Bsa 2uL
sample(extracted plasmid-size of vector 2.9kb, insert~4kb) 5uL
Enzyme (NotI) 1
total volume 20uL
The enzyme NotI will cut from the 3' end of the vector.
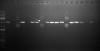
Lane1: 1kb ladder
Lane2: ctrl 1 (uncut extracted vector+insert)
Lane3: RE digest sample 1 (Not I)
Lane4: RE digest sample 2 (Not I)
Lane5: ctrl 2 (uncut extracted vector+insert)
Lane6: RE digest sample 3 (Not I)
Lane7: RE digest sample 4 (Not I)
Lane8: ctrl 3 (uncut extracted vecrot+insert)
Thanks in advance for the suggestion and opinion.
cheerioet on Oct 5 2009, 10:04 PM said:
perneseblue on Oct 1 2009, 06:35 PM said:
Could you write out your the restriction digest formulation that you used. How much of each component did you add, and at what temperature. How much DNA are you cutting? (Might you have accidentally used TE to make up the digestion cocktail)
NOTE:: Your restriction enzyme comes a glycerol based buffer. Glycerol in is inhibitor of enzymatic activity. The total volume of enzyme should not exceed 5% of the digest volume, otherwise you would start experiencing enzyme inhibition and star activity. In other words more is not better for a restriction digest.
Could you also tell us what size fragments that you expect from each restriction digest? And where it cuts in your vector-insert molecule.
You might also what to clean up your DNA again.
Should a DNA sample be dirty, (ie containing one of many enzyme inhibitory compounds) the DNA won't cut. In such an event, the DNA sample needs to be cleaned. A phenol/chloroform extraction would do the trick.
Dear Perneseblue,
Thanks for the fast reply. I am going to try a phenol/chloroform clean up before continuing the 2nd digestion.
I am using the followings protocol and formula.
dH2O 10uL
10X buffer3 2uL
10X Bsa 2uL
sample(extracted plasmid-size of vector 2.9kb, insert~4kb) 5uL
Enzyme (NotI) 1
total volume 20uL
The enzyme NotI will cut from the 3' end of the vector.
Lane1: 1kb ladder
Lane2: ctrl 1 (uncut extracted vector+insert)
Lane3: RE digest sample 1 (Not I)
Lane4: RE digest sample 2 (Not I)
Lane5: ctrl 2 (uncut extracted vector+insert)
Lane6: RE digest sample 3 (Not I)
Lane7: RE digest sample 4 (Not I)
Lane8: ctrl 3 (uncut extracted vecrot+insert)
Thanks in advance for the suggestion and opinion.
Opss...the gel image attached here.
thanks.
It appears to me that the digest did work. If you are expecting two DNA fragments from a NotI digest, then only Sample 3 might have your insert present. The others linearise fine and probably are empty vectors. You might want to try destaining your gel or digesting more plasmid DNA to get a brighter signal.
perneseblue on Oct 6 2009, 02:40 PM said:
Dear Perneseblue,
I am going to do a phenol/chloroform purification to continue with a second digestion and will try to see how it goes.
I am not sure if the empty vectors applicable for this case, because I am using a clean cut vector whereby when i try to religate the vector to propagate it, I didn't manage to do it successfully. Instead, when I am using a control positive insert to transform with this vector, there are growth of colonies and when I ran a gel electrophoresis to check on it(the uncut vector+insert), it shows the correct band size.
But I did not manage to run a gel of the insert+vector, together with a linearised vector; because of lacking of the linearised vector.
Thanks.
Cheerioet.

cheerioet on Oct 7 2009, 02:38 PM said:
perneseblue on Oct 6 2009, 02:40 PM said:
Dear Perneseblue,
I am going to do a phenol/chloroform purification to continue with a second digestion and will try to see how it goes.
I am not sure if the empty vectors applicable for this case, because I am using a clean cut vector whereby when i try to religate the vector to propagate it, I didn't manage to do it successfully. Instead, when I am using a control positive insert to transform with this vector, there are growth of colonies and when I ran a gel electrophoresis to check on it(the uncut vector+insert), it shows the correct band size.
But I did not manage to run a gel of the insert+vector, together with a linearised vector; because of lacking of the linearised vector.
Thanks.
Cheerioet.
Dear Perneseblue
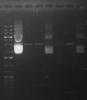
I had tried phenol/chloroform clean up for the RE digest. The last four lane from right were sequentially digest by another set of RE(s)- EcorI and HindIII. Yet, I did not see anything as in 2 bands cut. Do you think the originally extracted plasmid vector carried the insert? Lane 7 from right is the undigest extracted plasmid vector.
I really ran out of idea on how to trouble shoot this. More over, I don't have clean cut vector to run as a ctrl lane, as the vector came from a kit which has limited volume. :'(
I am really desperately need some help and options.
Thanks in advance for your advice.
Cheerioet