Mysterious PCR Contamination - (Sep/02/2009 )
Can you tell me what is going on?
Summary of project to date:
I am running a fairly standard amplification of bacterial DNA using a proven primer set (470bp amplicon).
Early amplifications from samples worked well, but a faint smearing with repetitive bands of fluorescence began to appear in later amplifications. This phenomenon progressively got brighter and more pronounced. No re-amplifications, just amplifications of various samples done over a period of a few weeks. The problem was first noted when the negatives failed to run clear.
Eventually, the bright smearing showed as a continuous line through any and all samples run, new or old, although the presence of the target amplicon still shows up as a discernible band through the smear.
At this point all PCR master mix reagents have been changed to fresh aliquots from clean master stocks. None of the changes had any effect. The samples are not the problem since the negatives appear contaminated. Primer dimer is notably absent. Note that the problem only occurs after amplification, and only with this particular primer set.
Attached is a photo from a recent troubleshooting experiment. This experiment tested a few variations to try and rule out problems with the polymerase/master mix being used.
Four different amplifications were run:
A) used a Econotaq master mix with a primer set for human genomic DNA (no amplicon expected); B is the same master mix with the target primer set; C) is the same as 'B' with a different batch of Econotaq (just in case); and finally D) a master mix prepared with the target primers and a PCR-ready bead instead of Econotaq.
Well 1 is the marker, 2-5 show the negatives; A, B, C, D respectively. 6 is a blank well. 7-10 show the positives, ordered as before.
If you tell me what is going on, I will buy you a beer next time you are in Indiana.
If your current primer stock is old, you might need to re-synthesize this pair of primers.
If I didn't know any better, I would have said you posted a gel showing various digestions of genomic DNA. How much gDNA did you add to each lane?
If the primers had gone off, as asword suggested, I would not have expected to see the higher MWt smear.
I hate to embarrass or offend you, but are you sure you added Taq polymerase, and not TaqI restriction endonuclease? Both would survive the temperature cycling, and TaqI would make a smear of gDNA...
I'd also be tempted to contact the company and see if they have any explanation. We have all heard of bad batches of enzymes.
Thank you for your suggestions,
I hope there is a simple solution, but none of these quite fit.
I have run the experiment with a newly synthesized primer set and an the older (year old) primer set with the same results.
I am definitely using econotaq (polymerase). My aliquots have all come from the working master stock used by several other researchers. As collumn A shows, totally normal results occur if I use a different primer.
The primer set (and econotaq, and DNA free water) worked fine until recently when the problems began.
Could your pipette barrel be contaminated?
I have seen something like this when I was working with environmental samples. We would get the expected product but also this unbelievable smear.
I need to think back about what we did. I may need to contact some old colleagues as well.
I might suggest that you dilute your DNA 1:20 and 1:100. See if it is an inhibitor that is playing in the reaction.
I will post again if I recall anything new.
We just did three amplifications under the cleaned hood, all new reagents, sterile pipettors, no chance of aerosol DNA. One amplification used a PCR-ready bead, the other two used Econotaq. Same problem.
Note that all three negatives show smearing, so too much template DNA should NOT be the problem--there is no template DNA in the negatives.
See attached image.
It is also not procedural (multiple researchers have run amplifications from start to finish now). , All the reagents are used with other primers with no problem. If there is contamination, it is contamination only picked up by my specific primer-set, but said contamination is highly unlikely to be in the Polymerase mix or water (Have been changed multiple times for fresh aliquots from different master stocks). It seems highly unlikely that two separately synthesized batches of primer would have the same contamination problem.
Most perplexing is that it worked at first. That suggests the primers were fine--if they went bad, the fresh primer set should have worked, at least for a little while.
Our thermocycler seems to be working fine with everyone else's research. We reprogrammed the protocols for my particular amplification in case something had gone wonkly there.
We are stumped. I am going to try having another company synthesize a fresh batch of my primers, I can try switching to yet another master stock of DNA-free water, and otherwise I have no idea. 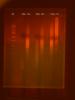
could your loading buffer be contaminated??
Hi,
I personally feel that is the problem comes on when you reconstitute your primers after you bought it from manufacturer. I guess is your DNA-free water problem. I wonder how you treat your water to make it DNA-free.
Your bottles that you use to store your miliQ water might have contaminated with other cells or bacteria and is not thoroughly clean, it might happen to be the bottle cap that you forget to clean. I suggest you get a nuclease free water, eg promega nuclease free water (i use it, no issues), and reconstitute your primers using that.
If your primers had been pre-constituted by manufacturer, try request for a freeze dry package.
Hope this helps.
 good luck
good luck
First of all, I think we can say there is no contamination in the reaction water, enzyme or dNTPs, since the tubes with human primers show nothing.
That only leaves the bacterial-specific primers.
Imagine this scenario. The primers start off OK, but with repeated freeze-thaws, start to degrade, from the 3' end (If the degradation was from the 5' end, you'd be less likely to bind to as many sites as you see, I think). The freeze-thawing could have been from the freezer (I presume it's not a frost-free freezer), or repeated removal to aliquot working stock. As more of the 3' end was degraded, the primers start mispriming more and more, hence the smear. Note that this mispriming must happen at a slow rate, as the expected band is seen as well as the smear.
Here's my suggestion: try a touchdown PCR. If primer degradation is the cause, you should see the smear reduce. Also a question: how many cycles do you usually do?