2-D Agarose Gel Electrophoresis of DNA Replication Intermediates
Bonita J. Brewer, October 28, 1991
For an in-depth review of the method, see Friedman, K. and Brewer, B. (1995) Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Meth. Enzymol. 262: 613-627.
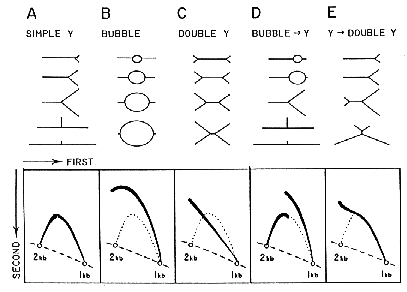
Two dimensional agarose gel electrophoresis is adapted from the procedure by Bell and Byers (Anal. Biochem. 130:527, 1983). The first dimension gel is intentionally run at low voltage in low percentage agarose to separate DNA molecules in proportion to their mass. The second dimension is run at high voltage in a gel of higher agarose concentration in the presence of Ethidium bromide so that the mobility of a non-linear molecule is drastically influenced by its shape. See the figure for interpretations of 2-D gel patterns obtained for different replication intermediates.
First Dimension: 0.4% agarose in 1XTBE (0.089M Tris-borate, 0.089 M Boric Acid, 0.002 M EDTA); electrophoresis buffer, 1XTBE.
Pour agarose in horizontal slab gel apparatus with a comb ~1.5 mm from the bottom support plate. The dimensions of the slab are somewhat arbitrary; convenient dimensions are 13 cm wide, 20 cm long, and ~0.6 cm thick. Use a comb with narrow, thin teeth (for example, 4 mm wide and 1.2 mm thick) to produce a narrow first dimension gel lane with tight, sharp bands. Load alternate lanes of the gel. The empty lanes prevent accidental cross-contamination when the lanes are excised for the second dimension. Carry out the electrophoresis, with the gel submerged by a few mm, at 0.7 to 1.0 V/cm for 15 to 30 hr. (If a voltmeter is not available, estimate the voltage required by measuring the distance between the electrodes of the gel box and set the voltage accordingly.) The exact conditions depend upon the size of the molecules to be analyzed. The band containing the fragment of interest and the band of twice that size should be separated by 3-5 cm in the first dimension gel. For example, a 3 kb fragment can be successfully separated from a 6 kb fragment by electrophoresis at 1V/cm for 15 to 18 hr. To achieve the same separation for a 6 kb fragment and a 12 kb fragment might require 24-30 hr at the same or lower voltage. It becomes appropriate to lower the voltage when large molecules are being examined since branched intermediates in this size range are more likely to retarded in their mobility. Thus they will not run at their true molecular weight. Krysan and Calos (1991, MCB 11; 1464-1472) have modified conditions for examining molecules larger than 6 kb.
Stain the first dimension slab gel in 1XTBE with 0.3 µg/ml ethidium bromide for ~20 min. Examine/photograph the gel on a long wave UV light box and mark lanes to be run in the second dimension. (Do not expose the gel to excessive doses of UV light as the DNA will become nicked and replicating structures can be lost.) Excise the lanes of interest with a clean razor blade--making at least one edge very straight, vertical, and close to the lane of interest. It is through this cut edge that the DNA will run in the second dimension. The lane can be trimmed at either end to include just that portion that contains the size range of interest. When possible, include the entire lane from the well to at least 1cm beyond the band of interest.
Second Dimension: 1.0 to 1.2 % agarose in 1XTBE and 0.3 µg/ml ethidium bromide; electrophoresis buffer is 1XTBE with 0.3 µg/ml ethidium bromide (2-3 L total) circulated from anode to cathode at 50 to 100 ml/min.
Place the lane on a clean gel support at 90o to the direction of electrophoresis. Convenient dimensions for the second gel are 10 cm wide (or the length of the first dimension gel lane) by 13 cm long. Permit the agarose to cool to ~55oC and then pour the agarose around the first dimension lane, to a final depth that just barely covers the first dimension gel lane. Submerge the gel in an electrophoresis tank (I have found that the apparatus sold by BRL, Model H1, works well. It has a large buffer capacity and the size of the gel platforms (20 cm wide, 25 cm long) will accomodate four 2-D gels that are 10X12.5 cm.) Perform the electrophoresis at 5 V/cm in a cold room (4oC) for 4-8 hr (depending on the size range of the molecules being analyzed). Check the progress of electrophoresis with a hand-held UV lamp. The largest molecules should run ~1/3 of the length of the gel with the smallest molecules near the bottom corner of the gel. After electrophoresis the gel can be photographed and treated as any gel for Southern analysis. Include the HCl treatment to facilitate transfer of large and branched molecules.
Additional hints:
Beware that some restriction enzymes have non-specific single-stranded nucleases as contaminants. These will destroy branched molecules. Check with manufacturer, or incubate the enzymes with single-stranded circles of M13 or PhiX174 DNA and assay on gels for breakage or degredation.
Avoid using spermidine in restriction enzyme digestions. Spermidine remains associated with the DNA during electrophoresis and reduces the charge on the DNA. The result is that a proportion of the fragments trail during the first dimension electrophoresis. In the second dimension the retarded DNA produces horizontal streaks across the gel.
Manipulating the 0.4% agarose gel slab can be tricky since it has so little gel strength and is so very slippery. Handle it with plastic support sheets. I have found the easiest way to manipulate the excised gel lanes is to scoop them up with a flexible plastic ruler.
Return to top of page
Go to fork-direction gel method