Either too low or too high a260/a230 - (Jan/12/2012 )
Hi,
For around a month I am trying to isolate RNA from leaf samples. The amount of my samples are not very much. I used both trizol method and qiagen Rneasy kit. In trizol, I always obtain peaks at a230, no matter how long do I wait at ethanol drying, how careful do I behave at obtaining upper phase etc. In Rneasy, I have no such problem but I can't obtain good concentration of samples, and today I even obtained a260/a230 ratio at around 4.5. If I do RNA cleanup to the samples I obtained from Trizol, this time the concentration of my RNA drops dramatically. What do you suggest me to do in order to obtain pure RNA at high concentration? In the attachments you see some of my results.
Thanks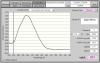
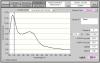
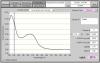
You likely have Trizol carry over or some other contaminate absorbing at 230, but also artificially inflating the concentration so that when you remove them, youre left with the actual amount of RNA....the super high ratio is weird...I did a microarray with sample that had a 260/230 of 2.5...the core people told me they didnt know what could make it high like that, but they recommended doing the spec reading in a buffered solution like TE vs water. I always use whatever the RNA is in..water or elution buffer.
Regarding trizol, you can do the phase separation for longer with the centrifuge at 4 degrees. Make sure the stay well away from the organic and aqueous bilayer when you do the chloroform extraction. Once you get the RNA, I love lithium chloride preciptiation for clean ups...it purifys RNA beautifully and always solves the issue when I have a low 260/230. It selectively precipitates RNA, (not proteins, DNA (for the most part) or Trizol). Kits that use Silica based columns are also great but a lot more expensive than trizol. I prefer RNAaqueous over RNeasy.
If concentration is an issue, bring it up in less volume...or, if its for real time, there are cDNA kits that work better for low inputs (Ive made cDNA from 200ng of RNA with no trouble, which worked great for real time).
Thanks for the answer. I read somewhere that if the concentration is low, you may have very high 260/230 ratio. This may be the problem. In addition to that, I realized that yesterday I couldn't crush the leaf samples enough. I think this was the main reason for me to obtain low concentration at qiagen trial. Can you explain what you meant by that "
What species are your leaf samples from? Some plants do not work well with Trizol or RNeasy -- many will give poor 260/230 ratios due to high polysaccharide content. The guanidine buffers used in Trizol and RNeasy have a tendency to coprecipitate polysaccharides along with nucleic acids.
You can improve perfomance of these methods by using less tissue (for RNeasy, instead of 100 mg tissue/450ul buffer, use less than 50mg/450ul buffer). Also, with RNeasy, use buffer RLC instead of RLT (RLT is more likely to cause problems with plant tissues).
This method gives very good RNA for some species, but it hasn't been tested on difficult plants:
Bilgin, D.D., DeLucia, E.H., and Clough, S.J. (2009) A robust plant RNA isolation method suitable for Affymetrix GeneChip analysis and quantitative real-time RT-PCR. Nat. Protoc. 4, 333-340.
CTAB protocols are commonly used to deal with plants high in polysaccharides and they usually are inexpensive. I can give you a few references if you like.
Regarding using TE, the PH of buffer can have a small impact on the ratios, but I read a some and its generally only by 0.2-0.3 in either direct. It does seem that at low concentrations small changes are amplified. The curve in the first pic looks good, but you may want to try DNAse treatment or running them through the column to ensure there isnt DNA in the sample. The trizol extracted samples should look better after LiCl; I generally lose some (~15%). Although its recommended to incubate for 30mins at -20, I leave the RNA/LiCL at -20 overnight, then centrifuge at 14000rpm for 15min at 4 degrees. Im am not familiar with the
Although we work with animal tissues, there are some (like skeletal muscle) that are fibrous. We pulverize difficult/tough samples using liquid nitrogen and a tissue pulverizer (something like this: https://www.spectrumlabs.com/labware/Pulverizer.html). This works fabulous on leaves, Ive tried it on a lark :~). It turns them into a fine powder.
Good luck!
Thanks for the answers, and sorry for the late answer. I was on my final exams. Anyway, I obtained better RNAs by using Qiagen kit more carefully, as I thought I had failed at liquid nitrogen crushing. Now, I got some cDNAs (hopefully). I did run PCR on them using Ubi primers, and it is on gel now. If I manage to see bands different than NC, I can happily go to qRT-PCR step finally.